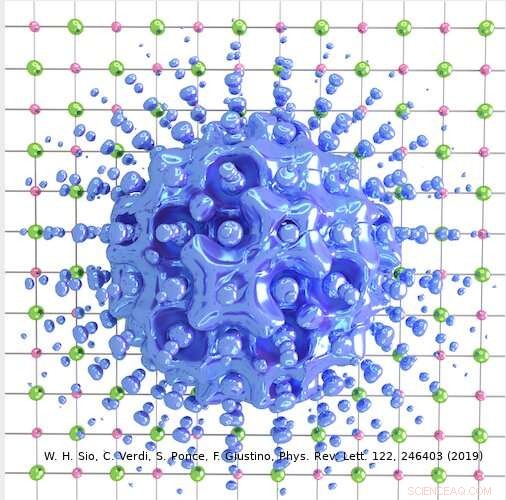
Upphovsman:Weng Hong Sio.
Ett team av forskare vid University of Oxford har nyligen introducerat ett nytt sätt att modellera polaroner, en kvasipartikel som vanligtvis används av fysiker för att förstå interaktioner mellan elektroner och atomer i fasta material. Deras metod, presenteras i ett papper publicerat i Fysiska granskningsbrev , kombinerar teoretisk modellering med beräkningssimuleringar, möjliggör fördjupade observationer av dessa kvasipartiklar i ett brett spektrum av material.
Väsentligen, en polaron är en sammansatt partikel som består av en elektron omgiven av ett moln av fononer (dvs. gittervibrationer). Denna kvasipartikel är tyngre än själva elektronen och på grund av sin betydande vikt kan den ibland fastna i ett kristallgitter.
Polaroner bidrar till den elektriska strömmen som driver flera tekniska verktyg, inklusive organiska ljusdioder och pekskärmar. Att förstå deras egenskaper är därför av största vikt, eftersom det kan hjälpa till att utveckla nästa generation av olika enheter för belysning och optoelektronik.
"Tidigare arbete med polaroner förlitade sig på idealiserade matematiska modeller, "Prof. Feliciano Giustino, chefen för teamet som genomförde studien, berättade för Phys.org. "Dessa modeller har varit mycket användbara för att förstå polarons grundläggande egenskaper, men de tar inte hänsyn till materialstrukturen i atomskala, därför är de inte tillräckliga när vi försöker studera verkliga material för praktiska tillämpningar. Vår idé var att utveckla en beräkningsmetodik som skulle möjliggöra systematiska undersökningar av polaroner med förutsägbar noggrannhet. "
Metoden som utarbetats av Giustinos team är baserad på densitet-funktionell teori, som för närvarande är det mest populära verktyget för modellering och design av prediktivt material med hjälp av kvantmekanik. En av de främsta utmaningarna när man studerar polaroner baserat på denna teori är att de nödvändiga beräkningsresurserna (CPU -timmarna) är proportionella mot den tredje effekten av antalet atomer som ska simuleras. Med andra ord, om man studerade två kristaller med 10 och 20 atomer per cell, beräkningen som krävs för att studera den andra kristallen skulle vara 8 gånger mer tidskrävande än den som krävs för den första.
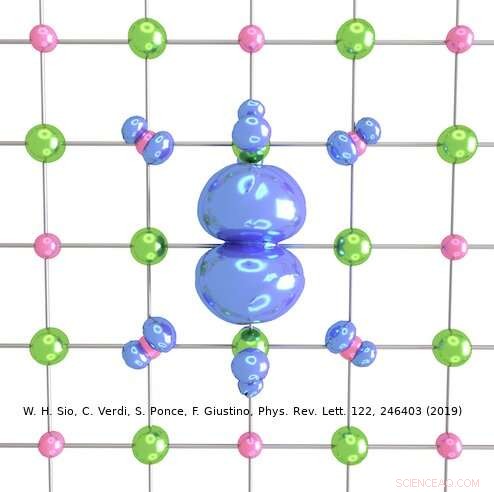
Upphovsman:Weng Hong Sio.
Eftersom många polaroner är 1-2 nanometer stora, beräkningar för att studera dessa system skulle kräva simuleringsceller med minst 3, 000-5, 000 atomer. Men nuvarande beräkningsmöjligheter skulle kämpa för att upprätthålla sådana simuleringar och var och en av de många beräkningar som krävs för att undersöka dessa system skulle ta veckor, även när du använder en modern superdator.
"Vår idé var att försöka göra denna process mer effektiv genom att utnyttja framsteg inom så kallad densitetsfunktionell störningsteori, "Weng Hong Sio, verkets första författare, förklarade. "Utan att gå in på detaljerna, vi kunde omarbeta problemet med att utföra en beräkning av ett polaron i en stor simuleringsceller till det enklare problemet att utföra flera beräkningar i den minsta enhetscellen i kristallen. Denna strategi öppnade nya möjligheter som tidigare var otillgängliga. "
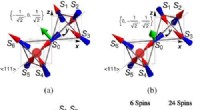
Den metod som utarbetats av Giustinos team kan användas för att beskriva både stora och små polaroner. I deras studie, till exempel, forskarna visade hur den kan användas för att beräkna vågfunktionerna, bildningsenergier och spektral nedbrytning av polaroner i LiF och Li 2 O 2 föreningar. Med hjälp av deras simuleringsmetod, de upptäckte att polaroner i enkla salter och metalloxider som används i batterier har en mycket rikare inre struktur än vad som föreslagits av tidigare arbeten inom området.
"Till exempel, i det prototypiska saltet litiumfluorid, man trodde tidigare att polaronen härrör från interaktionen mellan en elektron och longitidinala optiska fononer, dvs. gittervibrationerna som är ansvariga för kristallens dielektriska svar, "Förklarade Sio." Vi fann att dessa inte är de enda fononerna som är inblandade, och att interaktionen mellan elektronen och piezoakustiska fononer (dvs. vibrationerna som är ansvariga för piezoelektricitet) också är viktig. "
Observationerna som samlats in av Giustinos team förändrar det nuvarande perspektivet på polaronerna i salt litium fouride, vilket är ett mycket enkelt system. Att tillämpa deras metod på mer komplexa system kan avslöja ännu rikare strukturer, slutligen förbättra vår nuvarande förståelse för deras egenskaper och informera utvecklingen av nya material med skräddarsydda polatroniska egenskaper. I deras framtida forskning, forskarna planerar att använda sin metod för att studera annat material, för att ytterligare bedöma dess förutsägbarhet och uppnå en bättre förståelse för andra tekniskt viktiga material.
"Längre ner på linjen kommer det att vara viktigt att undersöka vad en polaron kan göra:för nu vet vi att vi kan beräkna den lägsta energikonfigurationen för ett polaron, men vi har ingen aning om vad som händer om denna polaron utsätts för statiska elektriska eller magnetiska fält eller för elektromagnetisk strålning, "Sa Giustino." Dessutom nära interaktioner med experimentella grupper kommer att vara avgörande för att översätta dessa resultat till applikationer. "
© 2019 Science X Network