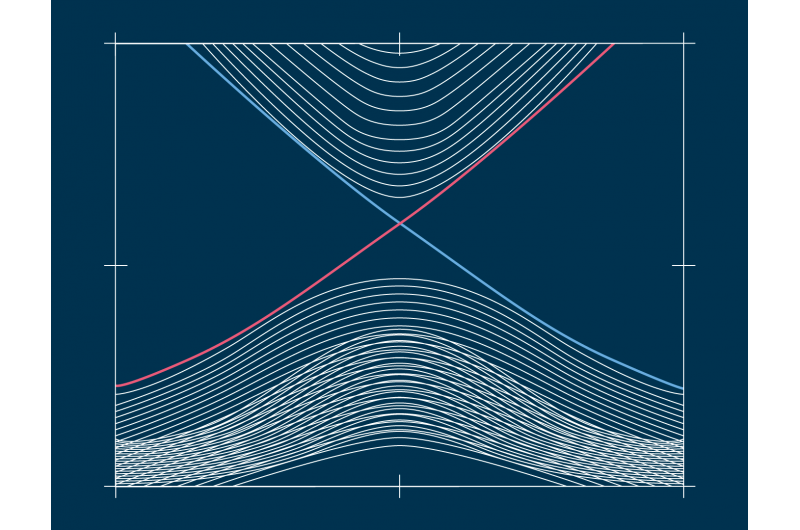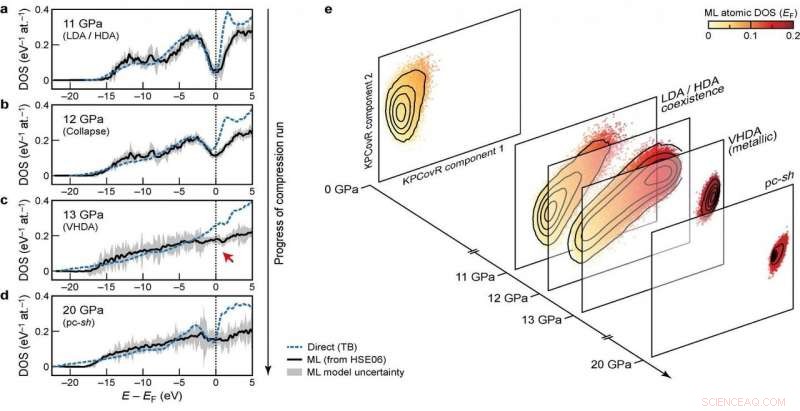
Elektroniska densiteter av tillstånd (DOS) i olika skeden av komprimeringskörningen Kredit:@Michele Ceriotti
Att kombinera elektroniska strukturberäkningar och teknik för maskininlärning (ML) har blivit ett vanligt tillvägagångssätt i atomistisk modellering av materia. Genom att använda de två teknikerna tillsammans har forskare, till exempel, att skapa modeller som använder atomkoordinater som de enda ingångarna för att på ett billigt sätt förutsäga alla egenskaper som kan beräknas med de första principberäkningarna som hade använts för att träna dem.
Medan de tidigaste och nu mest avancerade insatserna har fokuserat på att använda förutsägelser av totala energier och atomkrafter för att konstruera interatomiska potentialer, nyare ansträngningar har riktat sig till ytterligare egenskaper hos kristaller och molekyler såsom joniseringsenergier, NMR kemiska avskärmningar, dielektriska svarsegenskaper och laddningstäthet. I tidningen "Lär dig den elektroniska densiteten hos tillstånd i kondenserad materia, "Ceriotti och kollegor fokuserar på staternas elektroniska densitet (DOS), en annan mängd som ligger till grund för många användbara materialegenskaper, varav några kan observeras experimentellt.
DOS är i huvudsak antalet olika tillstånd som elektroner kan uppta vid en viss energinivå, och kan användas, till exempel, att beräkna det elektroniska bidraget till värmekapacitet i metaller och tätheten av gratis laddningsbärare i halvledare. Det är en indirekt proxy för egenskaper som energibandgapet, bandenergin och det optiska absorptionsspektrumet.
"Att förutsäga DOS är en intressant övning i sig eftersom det i huvudsak är den enklaste möjliga beskrivningen av den elektroniska strukturen bortom grundtillståndsbilden, "Sade Ceriotti." Det är också användbart eftersom det finns många egenskaper som du kan beräkna utifrån DOS, vilket gör det till ett bra exempel på hur nästa generation ML -modeller kan användas på liknande sätt som beräkningar av elektroniska strukturer, använda dem på ett indirekt sätt för att beräkna mellanliggande kvantiteter som sedan enkelt kan bearbetas för att utvärdera egenskaper som är svårare att lära sig direkt. "
Vid utvecklingen av modellen, gruppen försökte säkerställa överförbarhet över olika faser samt skalbarhet till stora systemstorlekar. Deras ultimata tillvägagångssätt, som tittar på hur olika atomkonfigurationer påverkar fördelningen av energinivåer, uppfyller dessa mål-den kunde lära sig och förutsäga DFT-beräknad DOS för en mångsidig datauppsättning av kiselstrukturer, täcker ett brett spektrum av termodynamiska förhållanden och olika faser. Det skala också linjärt, snarare än med kuben av antalet atomer som med elektroniska strukturberäkningar, gör det tillämpligt på stora strukturer. Till sist, modellen tillåter en analys av den lokala DOS, ger forskare chansen att undersöka samspelet mellan strukturella motiv och elektronisk struktur.
Kombinationen av överförbarhet, och skalbarhet av förutsägelser till stora systemstorlekar, göra modellen tillämplig för att ta itu med långvariga öppna frågor inom materialvetenskap. Det nya ramverket har redan använts för att belysa de elektroniska egenskaperna hos en 100 000-atomsimulering av amorft kisel, genomgår en serie fasövergångar när de komprimeras till 20 Gpa, i ett papper publicerat i Natur idag i samarbete med ett team bestående av forskare från Oxford, Cambridge, US Naval Research Laboratory och Ohio University. Den förutspådda DOS används också för att förklara hur de tryckinducerade strukturomvandlingarna kopplar ihop med materialets elektroniska struktur.
Att kombinera den nya modellen med en av de väletablerade potentiella energimodellerna gör det också möjligt att beräkna de elektroniska bidragen till makroskopiska egenskaper såsom metallers värmekapacitet och att utföra simuleringar som tar hänsyn till finite-elektroniska temperatureffekter-som visas i en annan snart publicerad artikel som diskuterar nickels högtemperaturegenskaper. Verkligen, den nya modellen är ett kritiskt steg mot MARVEL:s mål att utveckla integrerade maskininlärningsmodeller som ökar - och kanske så småningom ersätter - kostsamma elektroniska strukturberäkningar.
"Det finns andra egenskaper bortsett från elektrontätheten i tillstånd, såsom optiska excitationer, och NMR -svar, vilket vi har kunnat förutse exakt med maskininlärning. "sade Ceriotti." Om vi kan använda dem alla i kombination med billiga och exakta interatomiska potentialer kommer det att tillåta oss att beskriva alla egenskaper hos material med samma noggrannhet som uppnås med elektronisk strukturberäkning, men till en liten bråkdel av kostnaden. "