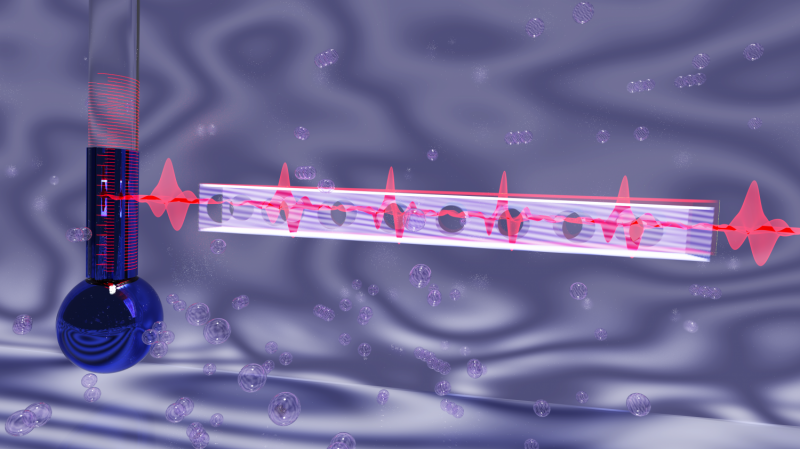
Automatiserad generering av datamängder ger ett mycket varierat urval av atompositioner för att träna en korrekt och allmän maskininlärningsmodell. Kredit:Los Alamos National Laboratory
En revolutionerande maskininlärningsmetod (ML) för att simulera atomers rörelser i material som aluminium beskrivs i veckans Naturkommunikation tidning. Detta automatiserade tillvägagångssätt för "interatomisk potentialutveckling" skulle kunna förändra området för upptäckt av beräkningsmaterial.
"Detta tillvägagångssätt lovar att vara en viktig byggsten för att studera materialskador och åldrande från första principer, " sade projektledaren Justin Smith från Los Alamos National Laboratory. "Simulering av dynamiken hos interagerande atomer är en hörnsten för att förstå och utveckla nya material. Maskininlärningsmetoder ger beräkningsvetenskapsmän nya verktyg för att exakt och effektivt genomföra dessa atomistiska simuleringar. Maskininlärningsmodeller som denna är designade för att efterlikna resultaten av mycket exakta kvantsimuleringar, till en liten bråkdel av beräkningskostnaden."
För att maximera den allmänna noggrannheten hos dessa maskininlärningsmodeller, han sa, det är viktigt att utforma en mycket mångsidig datauppsättning för att träna modellen. En utmaning är att det inte är självklart, a priori, vilka träningsdata som kommer att behövas mest av ML-modellen. Teamets senaste arbete presenterar en automatiserad metod för "aktivt lärande" för att iterativt bygga en träningsdatauppsättning.
Vid varje iteration, metoden använder den nuvarande bästa maskininlärningsmodellen för att utföra atomistiska simuleringar; när nya fysiska situationer uppstår som ligger utanför ML-modellens kunskap, nya referensdata samlas in via dyra kvantsimuleringar, och ML-modellen omskolas. Genom denna process, den aktiva inlärningsproceduren samlar in data om många olika typer av atomkonfigurationer, inklusive en mängd olika kristallstrukturer, och en mängd defektmönster som förekommer inom kristaller.