Inom kvantmekaniken kan partiklar existera i flera tillstånd samtidigt, vilket trotsar logiken i vardagliga upplevelser. Denna egenskap, känd som kvantsuperposition, är grunden för framväxande kvantteknologier som lovar att omvandla datoranvändning, kommunikation och avkänning. Men kvantöverlagringar står inför en betydande utmaning:kvantdekoherens. Under denna process bryts den känsliga överlagringen av kvanttillstånd ner när den interagerar med sin omgivande miljö.
För att låsa upp kemins kraft att bygga komplexa molekylära arkitekturer för praktiska kvanttillämpningar, måste forskare förstå och kontrollera kvantdekoherens så att de kan designa molekyler med specifika kvantkoherensegenskaper. Att göra det kräver att man vet hur man rationellt modifierar en molekyls kemiska struktur för att modulera eller mildra kvantdekoherens.
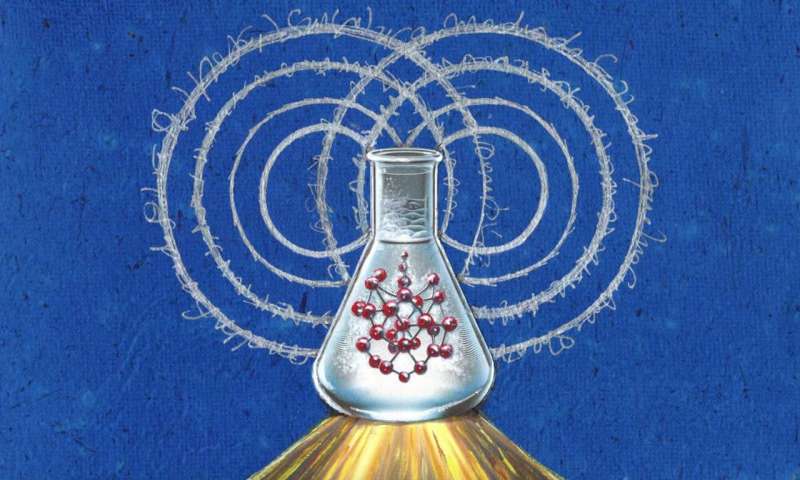
För detta ändamål behöver forskare känna till "spektraltätheten", den kvantitet som sammanfattar hur snabbt miljön rör sig och hur starkt den interagerar med kvantsystemet.
Fram till nu har kvantifiering av denna spektrala täthet på ett sätt som exakt återspeglar molekylernas krångligheter förblivit svårfångade för teori och experiment. Men ett team av forskare har utvecklat en metod för att extrahera den spektrala tätheten för molekyler i lösningsmedel med enkla Raman-resonansexperiment – en metod som fångar hela komplexiteten i kemiska miljöer.
Under ledning av Ignacio Franco, en docent i kemi och fysik vid University of Rochester, publicerade teamet sina resultat i Proceedings of the National Academy of Sciences .
Med hjälp av den extraherade spektrala tätheten är det möjligt att inte bara förstå hur snabbt dekoherensen sker utan också att bestämma vilken del av den kemiska miljön som är mest ansvarig för det. Som ett resultat kan forskare nu kartlägga dekoherensvägar för att koppla molekylstruktur med kvantdekoherens.
"Kemi bygger upp från idén att molekylstrukturen bestämmer materiens kemiska och fysikaliska egenskaper. Denna princip styr den moderna designen av molekyler för medicin, jordbruk och energitillämpningar. Med denna strategi kan vi äntligen börja utveckla kemiska designprinciper för framväxande kvantteknologier", säger Ignacio Gustin, doktorand i kemi vid Rochester och den första författaren till studien.
Genombrottet kom när teamet insåg att resonans-raman-experiment gav all information som behövdes för att studera dekoherens med full kemisk komplexitet. Sådana experiment används rutinmässigt för att undersöka fotofysik och fotokemi, men deras användbarhet för kvantdekoherens hade inte uppskattats.
De viktigaste insikterna framkom från diskussioner med David McCamant, docent vid kemiavdelningen i Rochester och expert på Ramanspektroskopi, och med Chang Woo Kim, nu på fakulteten vid Chonnam National University i Korea och expert på kvantdekoherens, medan han var postdoktor vid Rochester.
Teamet använde sin metod för att visa, för första gången, hur elektroniska superpositioner i tymin, en av byggstenarna i DNA, nyss upp på bara 30 femtosekunder (en femtosekund är en miljondel av en miljarddels sekund) efter dess absorption av UV ljus.
De fann att några vibrationer i molekylen dominerar de inledande stegen i dekoherensprocessen, medan lösningsmedel dominerar de senare stadierna. Dessutom upptäckte de att kemiska modifieringar av tymin avsevärt kan förändra dekoherenshastigheten, med vätebindningsinteraktioner nära tyminringen som leder till snabbare dekoherens.
I slutändan öppnar teamets forskning vägen mot att förstå de kemiska principerna som styr kvantdekoherens. "Vi är glada över att använda denna strategi för att äntligen förstå kvantdekoherens i molekyler med full kemisk komplexitet och använda den för att utveckla molekyler med robusta koherensegenskaper", säger Franco.
Mer information: Ignacio Gustin et al, Mapping electronic decoherence pathways in molecules, Proceedings of the National Academy of Sciences (2023). DOI:10.1073/pnas.2309987120
Journalinformation: Proceedings of the National Academy of Sciences
Tillhandahålls av University of Rochester