Material består av atomer, som är uppbyggda av protoner, neutroner och elektroner. Interaktionerna mellan dessa partiklar bestämmer materialets egenskaper, såsom dess styrka, ledningsförmåga och magnetiska beteende. Att förstå dessa interaktioner är avgörande för att designa nya material med önskade egenskaper för ett brett spektrum av applikationer, såsom energilagring, elektronik och katalys.
En av de mest exakta metoderna för att studera elektroners beteende i material är densitetsfunktionsteori (DFT), som är en allmänt använd metod för att beräkna den elektroniska strukturen hos atomer, molekyler och fasta ämnen. DFT-beräkningar kan dock vara beräkningsintensiva, särskilt för stora system eller de som innehåller tunga element, vilket gör dem utmanande att tillämpa i många praktiska fall.
Metoden med självständigt fält (SCF) involverar att lösa Kohn-Sham-ekvationerna, en uppsättning ekvationer som definierar DFT-beräkningar. I det traditionella tillvägagångssättet löses Kohn-Sham-ekvationerna genom att expandera elektronernas vågfunktioner i en ändlig uppsättning basfunktioner, såsom plana vågor. Detta tillvägagångssätt kan vara beräkningsmässigt dyrt, särskilt för system med ett stort antal atomer.
Den nya tekniken som utvecklats av Argonne-forskarna använder ett mer effektivt tillvägagångssätt som kallas planvågsbasuppsättningen. I detta tillvägagångssätt representeras vågfunktionerna på ett rutnät och projiceras sedan på en uppsättning plana vågor. Detta minskar beräkningskostnaderna för beräkningarna och gör det möjligt för forskare att studera större system med större noggrannhet och effektivitet.
"Utvecklingen av denna nya teknik är ett betydande genombrott inom området för beräkningsmaterialvetenskap", säger Dr. John Perdew, seniorforskare vid Argonne och en av studiens huvudutredare. "Det öppnar dörren till nya möjligheter för att studera elektronernas beteende i material, vilket kommer att påskynda utvecklingen av avancerade material."
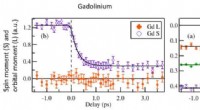
Forskarna visade kraften i sin nya teknik genom att studera en mängd olika material, inklusive kisel, vatten och ett komplext oxidmaterial. De fann att deras teknik kan uppnå liknande noggrannhet som traditionella DFT-beräkningar men med avsevärt minskade beräkningskostnader, vilket gör den till ett lovande verktyg för framtida materialforskning.
Studien, med titeln "Self-consistent field density functional theory with a planewave basis set:Formalism and implementation," publicerades i Journal of Chemical Physics och fick stöd av DOE Office of Science. Forskargruppen inkluderade forskare från Argonne National Laboratory, University of California, Berkeley och University of Illinois i Urbana-Champaign.