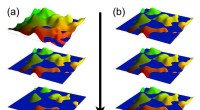
Schematisk skildring av olika energiterm som bidrar till adsorptionsenergin, och laddningstäthetsskillnad av 2H-P efter adsorption på Cu(111) vid 12,8 Ångström separation. Kredit:M. Müller/TU München
När vi fortsätter att krympa elektroniska komponenter, top-down tillverkningsmetoder börjar närma sig en fysisk gräns på nanoskala. Istället för att fortsätta att tappa bort vid denna gräns, en lösning av intresse innebär att man använder botten-upp självmontering av molekylära byggstenar för att bygga nanoskala enheter.
Framgångsrik självmontering är en noggrant koreograferad dans, där de attraktionskrafter och frånstötande krafter i molekyler, mellan varje molekyl och dess grannar, och mellan molekyler och ytan som stöder dem, måste alla beaktas. För att bättre förstå självmonteringsprocessen, forskare vid Münchens tekniska universitet har karakteriserat bidragen från alla interaktionskomponenter, såsom kovalent bindning och van der Waals-interaktioner mellan molekyler och mellan molekyler och en yta.
"I ett idealiskt fall, den minsta möjliga enheten har storleken på en enda atom eller molekyl, sa Katharina Diller, som arbetade som postdoktor i gruppen Karsten Reuter vid Münchens tekniska universitet. Reuter och hans kollegor presenterar sitt arbete denna vecka i Journal of Chemical Physics .
Ett sådant exempel är en enkel-porfyrin-switch, som upptar en yta på endast en kvadratnanometer. Porfinmolekylen, som var föremål för denna studie, är ännu mindre än så här. Porfyriner är en grupp av ringade kemiska föreningar som särskilt inkluderar hem - ansvarig för transport av syre och koldioxid i blodomloppet - och klorofyll. I syntetiskt härledda applikationer, porfyriner studeras för deras potentiella användningsområden som sensorer, ljuskänsliga färgämnen i organiska solceller, och molekylära magneter.
Forskarna från TU München bedömde interaktionerna mellan porfyrinmolekylen 2H-porfin genom att använda densitetsfunktionsteori, en kvantmekanisk beräkningsmodelleringsmetod som används för att beskriva de elektroniska egenskaperna hos molekyler och material. Deras simuleringar utfördes på den högpresterande superdatorn SuperMUC vid Leibniz-Rechenzentrum i Garching.
De metalliska substraten som forskarna valde för porfyrinmolekylerna att montera på, de tätt packade enkristallytorna av koppar och silver, används ofta som substrat inom ytvetenskap. Detta beror på ytornas tätt packade natur, som tillåter molekylerna att uppvisa en jämn adsorptionsmiljö. Dessutom, koppar och silver reagerar var och en med porhyriner - molekylen adsorberar starkare på koppar, medan silver gör ett bättre jobb med att hålla molekylens elektroniska struktur intakt - vilket gör det möjligt för forskarna att övervaka en mängd olika konkurrerande effekter för framtida tillämpningar.
I sin simulering, porfyrinmolekyler placerades på en koppar- eller silverplatta, som upprepades periodiskt för att simulera en förlängd yta. Efter att ha hittat den optimala geometrin i vilken molekylerna skulle adsorbera på ytan, forskarna ändrade storleken på metallplattan för att öka eller minska avståndet mellan molekyler, simulerar således olika molekylära täckningar. Beräkningsinställningen gav dem en switch för att slå på och av energibidragen från närliggande molekyler, för att observera samspelet mellan de enskilda interaktionerna.
Diller och Reuter, tillsammans med kollegorna Reinhard Maurer och Moritz Müller, vem är första författare på tidningen, fann att de svaga långväga van der Waals-interaktionerna gav det största bidraget till interaktionen mellan molekyl och yta, och visade att de ofta använda metoderna för att kvantifiera de elektroniska avgifterna i systemet måste användas med försiktighet. Förvånande, medan interaktioner direkt mellan molekyler är försumbara, forskaren hittade indikationer för ytmedierade molekyl-molekyl-interaktioner vid högre molekylär täckning.
"Analysen av den elektroniska strukturen och de individuella interaktionskomponenterna tillåter oss att bättre förstå självmonteringen av porfin adsorberat på koppar och silver, och möjliggör dessutom förutsägelser för mer komplexa porfyrinanaloger, "Diller sa. "Dessa slutsatser, dock, komma utan att ännu överväga effekterna av atomrörelse vid ändlig temperatur, som vi inte studerade i detta arbete. "