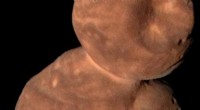
Maskininlärning förutsäger nanopartiklarnas struktur och dynamik Nanostrukturer som dessa tiolklädda guldnanopartiklar kan nu studeras med hjälp av den nya maskininlärningsmetoden som utvecklats vid Jyväskylä universitet. Metoden kan förutsäga den potentiella energin för en given struktur tillförlitligt. Kredit:Antti Pihlajamäki/Jyväskylä universitet
Forskare vid Nanoscience Center och vid fakulteten för informationsteknologi vid Jyväskylä universitet i Finland har visat att nya distansbaserade maskininlärningsmetoder som utvecklats vid universitetet i Jyväskylä kan förutse strukturer och atomdynamik hos nanopartiklar på ett tillförlitligt sätt. De nya metoderna är betydligt snabbare än traditionella simuleringsmetoder som används för nanopartikelforskning och kommer att underlätta mer effektiva undersökningar av partikel-partikelreaktioner och partiklarnas funktionalitet i sin miljö. Studien publicerades i en specialutgåva för maskininlärning i Journal of Physical Chemistry den 15 maj, 2020.
De nya metoderna tillämpades på ligandstabiliserade metallnanopartiklar, som länge har studerats vid Nanoscience Center vid universitetet i Jyväskylä. Förra året, forskarna publicerade en metod som framgångsrikt kan förutsäga bindningsställen för de stabiliserande ligandmolekylerna på nanopartikelytan. Nu, ett nytt verktyg skapades som på ett tillförlitligt sätt kan förutsäga potentiell energi baserat på partikelns atomstruktur, utan att behöva använda numeriskt tunga elektroniska strukturberäkningar. Verktyget underlättar Monte Carlo -simuleringar av partiklernas atodynamik vid förhöjda temperaturer.
Potentiell energi i ett system är en grundläggande mängd inom beräkningsnanovetenskap, eftersom det möjliggör kvantitativa utvärderingar av systemets stabilitet, hastigheter av kemiska reaktioner och styrkor av interatomiska bindningar. Ligandstabiliserade metallnanopartiklar har många typer av interatomiska bindningar med varierande kemisk styrka, och traditionellt har energiutvärderingarna gjorts med hjälp av den så kallade densitetsfunktionella teorin (DFT) som ofta resulterar i numeriskt tunga beräkningar som kräver användning av superdatorer. Detta har uteslutit effektiva simuleringar för att förstå nanopartiklarnas funktioner, t.ex., som katalysatorer, eller interaktioner med biologiska objekt som proteiner, virus, eller DNA. Maskininlärningsmetoder, en gång utbildad i att modellera systemen på ett tillförlitligt sätt, kan påskynda simuleringarna med flera storleksordningar.
Den nya metoden gjorde det möjligt att köra simuleringar på en bärbar eller stationär dator
I detta arbete använde forskarna de potentiella energierna, förutsägt av maskininlärningsmetoden, för att simulera atomdynamiken hos tiolstabiliserade guldnanopartiklar. Resultaten överensstämde väl med de simuleringar som utfördes med hjälp av densitetsteori. Den nya metoden gjorde det möjligt att köra simuleringar på en bärbar eller stationär dator i en tidsskala på några timmar medan referens -DFT -simuleringarna tog dagar i en superdator och samtidigt använde hundratals eller till och med tusentals datorkärnor. Hastigheten kommer att tillåta simuleringar av partiklarnas strukturella förändringar och partikel-partikelreaktioner vid förhöjda temperaturer under lång tid.
Forskarna använde en distansbaserad maskininlärningsmetod som utvecklats i gruppen av professor Tommi Kärkkäinen i Jyväskylä. Den beskriver varje momentan atomkonfiguration av en nanopartikel genom att beräkna en så kallad descriptor, och jämför avstånd mellan deskriptorer i ett flerdimensionellt numeriskt utrymme. Genom att använda korrelationer till en träningsuppsättning som skapats av referens -DFT -simuleringarna, den potentiella energin kan förutsägas. Detta tillvägagångssätt, används nu för första gången inom nanopartikelforskning, är enklare och mer transparent än traditionellt använda neurala nätverk.
"Det är oerhört motiverande att vi kan minska beräkningsbelastningen från att köra simuleringar i superdatorer till att köra dem med liknande kvalitet i en bärbar dator eller en hemdator, "säger doktoranden Antti Pihlajamäki som är huvudförfattare till studien.
"Det var en stor överraskning att våra relativt enkla maskininlärningsmetoder fungerar så bra för komplicerade nanostrukturer, "konstaterar professor Tommi Kärkkäinen.
"I nästa fas, vårt mål är att generalisera metoden för att fungera bra för nanopartiklar i många olika storlekar och kemiska kompositioner. Vi kommer fortfarande att behöva superdatorer för att generera tillräckligt med högkvalitativ data för att träna maskininlärningsalgoritmen, men vi hoppas att vi i framtiden kan flytta till att använda dessa nya metoder främst till studier av nanopartikelfunktionalitet i komplicerade kemiska miljöer, "sammanfattar akademiprofessorn Hannu Häkkinen, som samordnade studien.