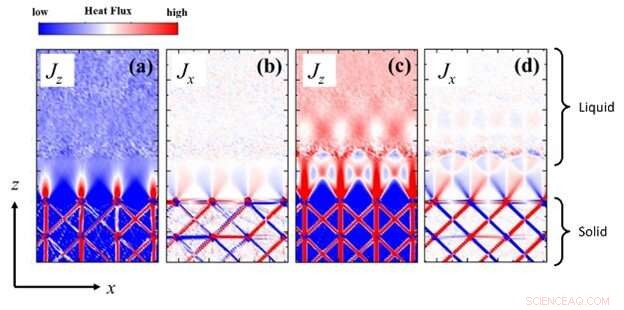
Fig. 1:Struktur av tvådimensionellt värmeflöde vid ett fast-vätskegränssnitt där temperaturgradienten är i z-riktningen, under (a,b) dåliga eller (c,d) goda vätbarhetsförhållanden. Simuleringsförhållandena skiljer sig från resultaten i uppsatsen. Kredit:Kunio Fujiwara och Masahiko Shibahara
Forskare vid Osaka University har simulerat värmetransport i de minsta skalorna med hjälp av en datorsimulering av molekylär dynamik. Genom att studera rörelserna hos de enskilda partiklarna som utgör gränsen mellan ett fast ämne och en vätska har de kunnat beräkna värmeflöde med oöverträffad precision. Detta arbete kan leda till betydande förbättringar i vår förmåga att tillverka enheter i nanoskala, såväl som funktionella ytor och nanofluidiska enheter.
Processen genom vilken värme överförs vid den punkt där ett fast ämne möter en vätska kan tyckas vara ett enkelt fysikproblem. Traditionellt användes makroskopiska storheter - såsom densitet, tryck, temperatur och värmekapacitet - för att beräkna hastigheten med vilken termisk energi rör sig mellan material. Att korrekt redogöra för individuella molekylers rörelse, samtidigt som man observerar lagarna för bevarande av energi och momentum, tillför en hel del komplexitet. Förbättrade datorsimuleringar i atomskala skulle vara ovärderliga för att mer exakt förstå ett brett spektrum av verkliga tillämpningar, särskilt inom området nanoteknik.
Nu har ett team av forskare vid Osaka University utvecklat en ny numerisk teknik för att visualisera ett modellerat värmeflöde på atomär skala för första gången. "För att i grunden förstå termisk transport genom ett fast-vätskegränssnitt måste transportegenskaperna hos atomer och molekyler beaktas", förklarar första författaren till studien Kunio Fujiwara. "Vi modellerade värmeflödet nära ett fast-vätskegränssnittsområde med subatomär rumslig upplösning genom att använda klassiska simuleringar av molekylär dynamik. Detta gjorde det möjligt för oss att skapa bilder av energiflödets tredimensionella struktur medan värme överfördes mellan lagren ."
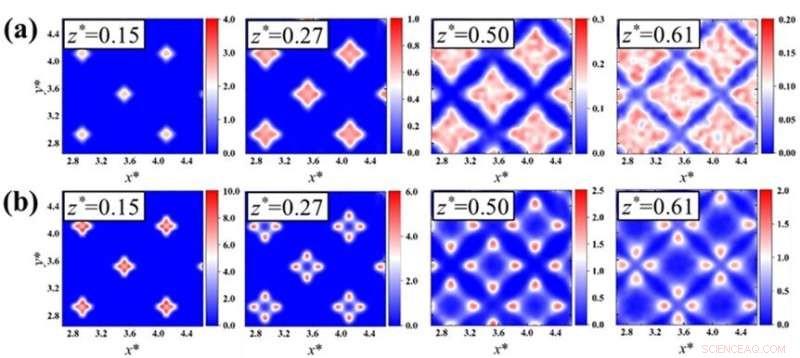
Fig. 2:Struktur av tredimensionellt värmeflöde vid ett fast-vätskegränssnitt vid specificerade z-ställen under (a,b) dåliga eller (c,d) goda vätbarhetsförhållanden. Simuleringsförhållandena skiljer sig från resultaten i papperet. Kredit:Kunio Fujiwara och Masahiko Shibahara
Med hjälp av den populära Lennard-Jones potentialen för att beräkna interaktionerna mellan intilliggande atomer fann teamet att värmeflödets riktning starkt beror på de subatomära spänningarna i strukturerna hos de fasta ämnena eller vätskorna.
"Förut fanns det inget bra sätt att visualisera värmeflöde i atomär skala", säger seniorförfattaren Masahiko Shibahara. "Dessa fynd borde göra det möjligt för oss att belysa och modifiera den termiska transporten baserat på 3D-värmeflödeskonfigurationen."
Detta kan göra det möjligt för skräddarsydd tillverkning i nanoskala att utföras mer effektivt. + Utforska vidare