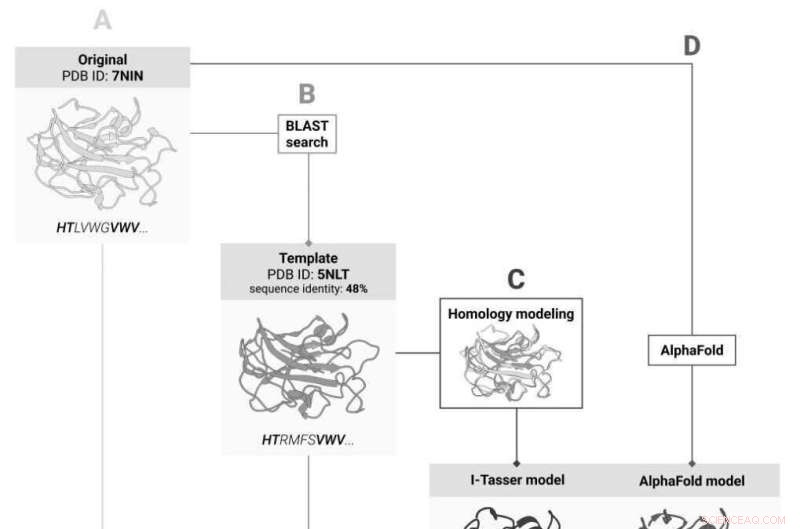
Fyra sätt att förutsäga förändringar i proteinstabilitet efter mutation:(A) genom strukturen av det ursprungliga proteinet; (B) genom strukturen av dess homolog; (C) av strukturen av det ursprungliga proteinet som förutsägs baserat på strukturen av holomlogen, och (D) av strukturen som förutsägs av artificiell intelligens baserat på aminosyrasekvensen. Kredit:Skolkovo Institute of Science and Technology
Forskare från Skoltech Center for Molecular and Cellular Biology jämförde olika metoder för förutsägelse av proteinstruktur när det gäller utvärdering av mutantproteinstabilitet och erhöll samma resultat för de AI-förutspådda strukturerna och experimentella tredimensionella (3D) av proteiner med liknande aminosyrasekvenser. Försöket att förutsäga målproteinets struktur från den kända strukturen hos dess "släkting" gjorde dock bara förutsägelsen mindre exakt. Teamets resultat kommer att underlätta preliminära beräkningar i utvärderingen av stabilitetsförändringar orsakade av mutation. Forskningen publicerades i Bioinformatics .
Biologiska experiment involverar ofta mutantproteiner, som behövs för att studera proteinstrukturen och funktionerna eller cellprocesserna, samt proteinteknik. Det är känt att mutationer påverkar ett proteins struktur och stabilitet. Eftersom experiment är för dyra och tidskrävande, skapar forskare en lösning i form av beräkningsmetoder för att utvärdera effekten av mutationer på stabiliteten. Deras tillämpningar kräver dock kunskap om ett proteins 3D-struktur.
En experimentell 3D-struktur är inte tillgänglig för alla proteiner och kommer sannolikt att saknas för den som teamet siktar på. Om så är fallet kan 3D-modeller av proteinets homologer, det vill säga dess "närmaste släktingar", ge livlinan, eftersom graden av likhet i aminosyrasekvenser som säkerställer en bra matchning mellan proteinernas 3D-strukturer är välkänd. Lösningen skulle vara att först förutsäga proteinets struktur baserat på den kända strukturen för dess homolog och sedan beräkna effekten av mutationer för den förutsagda modellen.
Tack vare förra årets genombrott i förutsägelse av proteinstruktur har forskarna nu ett alternativ:istället för att förutsäga 3D-strukturen baserat på homologer kan de använda det AI-baserade verktyget AlphaFold som förutsäger proteinstrukturen från aminosyrasekvensen och som redan har hanterat med de allra flesta proteiner som är kända till dags dato.
I sin senaste studie beslutade Skoltech-forskarna att ta reda på vilket av dessa tillvägagångssätt som fungerar bäst för att förutsäga stabilitetsförändringar vid mutation. Hur exakt AlphaFold än kan vara, är det fortfarande "guldstandarden" att hitta proteinstrukturen genom experiment. När de jämförde de två metoderna använde teamet sju stabilitetsutvärderingsmetoder och jämförde deras resultat med AlphaFold och I-Tasser, det bästa homologbaserade strukturförutsägelsesystemet. Forskarna kontrollerade också om de kan hoppa över den homologbaserade strukturförutsägelsen och beräkna stabiliteten för det homologa proteinets kända struktur.
"Vi bestämde oss för att ta reda på hur långt vi skulle avvika från korrekt förutsägelse om vi använde den "angränsande" proteinstrukturen istället för den riktiga. Det visade sig att det homologibaserade förutsägelsesteget bara gör saken värre genom att ge ett mindre exakt resultat. Vi har visat att det praktiskt taget inte spelar någon roll om du använder homologens experimentella struktur eller AlphaFolds förutsägelse.På sätt och vis handlade det här om validering:när du står inför en ny metod måste du kontrollera om den fungerar för din uppgift i första hand . Det är precis vad vi gjorde," första författaren till studien, Skoltech Ph.D. student Marina Pak, kommenterar.
"Med allt detta krångel över AlphaFold tror vissa forskare och icke-professionella att det har löst alla proteinforskningsfrågor inom beräkningsbiologi, men det har det inte. Till exempel visar förutsägelsen av mutationsinducerade stabilitetsförändringar fortfarande ganska låg tillförlitlighet, till och med även om förändringen i stabilitet är en av de viktigaste drivkrafterna för proteinfunktionalitet. Ett verktyg som entydigt skulle kunna bestämma effekten av mutationer på stabiliteten skulle hjälpa både att planera experimentet och tolka resultaten. Antag att för ett protein som inte är optimalt i termer av stabilitet vill vi hitta mutationer som skulle göra det stabilt under de önskade förhållandena, till exempel säkerställa att det förblir aktivt vid hög temperatur. När vi väl kan göra detta genom beräkningar enbart kommer tillvägagångssättet för proteinomformning och optimering att förändras dramatiskt." huvudförfattare till studien, avslutar Skoltech biträdande professor Dmitry Ivankov.
Även om förutsägelse av stabilitetsförändringar verkar lättare än att förutsäga 3D-strukturen, är det fortfarande en svårlöst utmaning även för AI. Knappa träningsdata är bara ett av problemen:AlphaFold hade nästan 200 000 proteinstrukturer att träna, men experimentella data om stabilitetsförändringar uppgår till tusentals set samtidigt som de täcker bara några dussin unika proteiner. Författarna hoppas att om mer data blir tillgänglig och forskare visar ett större intresse för uppgiften, kommer ett genombrott säkert att ske snart. + Utforska vidare