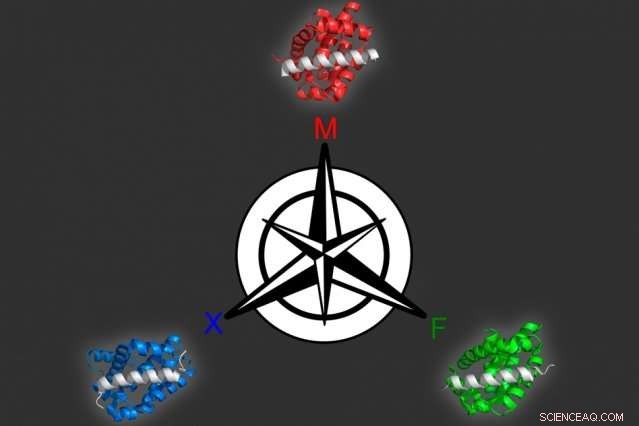
Genom att använda en datormodelleringsmetod som de utvecklade, MIT-biologer identifierade tre olika proteiner som kan binda selektivt till vart och ett av tre liknande mål, alla medlemmar av Bcl-2-familjen av proteiner. Kredit:Vincent Xue
Att designa syntetiska proteiner som kan fungera som läkemedel mot cancer eller andra sjukdomar kan vara en tråkig process:Det innebär i allmänhet att skapa ett bibliotek med miljontals proteiner, screenar sedan biblioteket för att hitta proteiner som binder till rätt mål.
MIT-biologer har nu kommit med ett mer förfinat tillvägagångssätt där de använder datormodellering för att förutsäga hur olika proteinsekvenser kommer att interagera med målet. Denna strategi genererar ett större antal kandidater och ger också större kontroll över en mängd olika proteinegenskaper, säger Amy Keating, en professor i biologi och biologisk teknik och ledare för forskargruppen.
"Vår metod ger dig en mycket större spelplan där du kan välja lösningar som skiljer sig mycket från varandra och kommer att ha olika styrkor och förpliktelser, " säger hon. "Vår förhoppning är att vi kan tillhandahålla ett bredare utbud av möjliga lösningar för att öka genomströmningen av de första träffarna till användbara, funktionella molekyler."
I en tidning som visas i Proceedings of the National Academy of Sciences veckan den 15 oktober, Keating och hennes kollegor använde detta tillvägagångssätt för att generera flera peptider som kan rikta sig mot olika medlemmar av en proteinfamilj som kallas Bcl-2, som hjälper till att driva cancertillväxt.
Nyligen doktorsmottagarna Justin Jenson och Vincent Xue är huvudförfattarna till uppsatsen. Andra författare är postdoc Tirtha Mandal, tidigare labbtekniker Lindsey Stretz, och tidigare postdoc Lothar Reich.
Modellerande interaktioner
Proteinläkemedel, kallas även bioläkemedel, är en snabbt växande klass av läkemedel som lovar att behandla ett brett spektrum av sjukdomar. Den vanliga metoden för att identifiera sådana läkemedel är att screena miljontals proteiner, antingen slumpmässigt utvalda eller utvalda genom att skapa varianter av proteinsekvenser som redan visat sig vara lovande kandidater. Detta involverar ingenjörsvirus eller jäst för att producera vart och ett av proteinerna, sedan exponera dem för målet för att se vilka som binder bäst.
"Det är standardmetoden:Antingen helt slumpmässigt, eller med vissa förkunskaper, designa ett bibliotek av proteiner, och sedan fiska i biblioteket för att dra ut de mest lovande medlemmarna, " säger Keating.
Även om den metoden fungerar bra, den producerar vanligtvis proteiner som är optimerade för endast en enskild egenskap:hur väl den binder till målet. Det tillåter inte någon kontroll över andra funktioner som kan vara användbara, såsom egenskaper som bidrar till ett proteins förmåga att ta sig in i celler eller dess tendens att provocera fram ett immunsvar.
"Det finns inget självklart sätt att göra den typen av saker - specificera en positivt laddad peptid, till exempel – med hjälp av screening av brute force-biblioteket, " säger Keating.
En annan önskvärd egenskap är förmågan att identifiera proteiner som binder hårt till sitt mål men inte till liknande mål, vilket bidrar till att läkemedel inte får oavsiktliga biverkningar. Standardmetoden tillåter forskare att göra detta, men experimenten blir mer besvärliga, säger Keating.
Den nya strategin innebär att först skapa en datormodell som kan relatera peptidsekvenser till deras bindningsaffinitet för målproteinet. För att skapa denna modell, forskarna valde först ut cirka 10, 000 peptider, vardera 23 aminosyror långa och spiralformade i struktur, och testade deras bindning till tre olika medlemmar av Bcl-2-familjen. De valde avsiktligt några sekvenser som de redan visste skulle binda bra, plus att andra de visste inte skulle göra det, så modellen kan inkludera data om en rad bindningsförmågor.
Från denna uppsättning data, modellen kan producera ett "landskap" av hur varje peptidsekvens interagerar med varje mål. Forskarna kan sedan använda modellen för att förutsäga hur andra sekvenser kommer att interagera med målen, och generera peptider som uppfyller de önskade kriterierna.
Med den här modellen, forskarna producerade 36 peptider som förutspåddes binda hårt till en familjemedlem men inte de andra två. Alla kandidater presterade extremt bra när forskarna testade dem experimentellt, så de försökte ett svårare problem:att identifiera proteiner som binder till två av medlemmarna men inte den tredje. Många av dessa proteiner var också framgångsrika.
"Det här tillvägagångssättet representerar en förändring från att ställa ett mycket specifikt problem och sedan designa ett experiment för att lösa det, att investera lite arbete i förväg för att skapa detta landskap av hur sekvens är relaterad till funktion, fånga landskapet i en modell, och sedan kunna utforska det efter behag för flera fastigheter, " säger Keating.
Sagar Khare, en docent i kemi och kemisk biologi vid Rutgers University, säger att den nya metoden är imponerande i sin förmåga att skilja mellan närbesläktade proteinmål.
"Selektivitet av droger är avgörande för att minimera effekter utanför målet, och ofta är selektivitet mycket svår att koda eftersom det finns så många liknande molekylära konkurrenter som också kommer att binda läkemedlet förutom det avsedda målet. Detta arbete visar hur man kodar denna selektivitet i själva designen, säger Khare, som inte var involverad i forskningen. "Tillämpningar i utvecklingen av terapeutiska peptider kommer nästan säkert att följa."
Selektiva droger
Medlemmar av Bcl-2-proteinfamiljen spelar en viktig roll för att reglera programmerad celldöd. Dysreglering av dessa proteiner kan hämma celldöd, hjälpa tumörer att växa okontrollerat, så många läkemedelsföretag har arbetat med att utveckla läkemedel som riktar sig till denna proteinfamilj. För att sådana läkemedel ska vara effektiva, det kan vara viktigt för dem att rikta in sig på bara ett av proteinerna, eftersom att störa dem alla kan orsaka skadliga biverkningar i friska celler.
"I många fall, cancerceller verkar använda bara en eller två medlemmar av familjen för att främja cellöverlevnad, " säger Keating. "I allmänhet, det är erkänt att det skulle vara mycket bättre att ha en panel av selektiva agenter än ett grovt verktyg som bara slog ut dem alla."
Forskarna har ansökt om patent på peptiderna de identifierade i denna studie, och de hoppas att de kommer att testas ytterligare som möjliga droger. Keatings labb arbetar nu med att tillämpa denna nya modelleringsmetod på andra proteinmål. Denna typ av modellering kan vara användbar för att inte bara utveckla potentiella läkemedel, men också att generera proteiner för användning i jordbruks- eller energitillämpningar, hon säger.
Den här historien återpubliceras med tillstånd av MIT News (web.mit.edu/newsoffice/), en populär webbplats som täcker nyheter om MIT-forskning, innovation och undervisning.