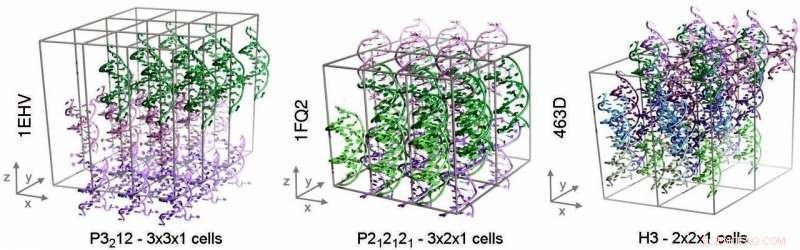
De tre kristallsystemen som undersöktes i denna studie. Dessa system innehåller (från vänster till höger) 27, 24 respektive 36 dubbelsträngat DNA. Kredit:Pablo Dans Puiggròs, IRB Barcelona
Sedan strukturbiologins födelse, Röntgenkristallografi har varit den mest använda tekniken för att bestämma den tredimensionella strukturen hos biomolekyler, de kemiska föreningar som finns i levande organismer. I detta avseende kunskap om interaktionerna mellan biomolekylerna med deras kristallmiljö och de molekylära krafterna som stabiliserar kristallerna skulle tjäna till att optimera denna teknik.
En studie publicerad i tidskriften Chem och utförd av forskare vid Institutet för forskning i biomedicin (IRB Barcelona) är den första som uppnår stabila simuleringar av DNA-kristaller. Denna prestation har gjort det möjligt för forskarna att förklara vikten av de kemiska tillsatser som används experimentellt för att uppnå lämpliga kristallisationsförhållanden och stabila kristaller i laboratoriet.
"Den första att dra nytta av denna studie är gemenskapen av biofysiker och beräkningsfysiker/kemister, som nu har en referens och tydliga protokoll för att uppnå stabila simuleringar av DNA-kristaller, " säger Pablo D. Dans, postdoktor vid IRB Barcelona.
Leds av Modesto Orozco, chef för laboratoriet för molekylär modellering och bioinformatik, studien presenterar den mest detaljerade atombeskrivningen av egenskaperna hos DNA-kristaller hittills.
"I längden, simuleringen av flera kristaller som erhållits under en rad experimentella förhållanden bör göra det möjligt för oss att förutse och förutsäga effekten av en given kemisk tillsats, vilket tjänar till att vägleda kristallografer i deras experiment och avsevärt minska kostnaden och tiden som behövs för att erhålla kristallerna, " säger Modesto Orozco, vars labb är en internationell referens inom bimolekylär beräkning och simulering.