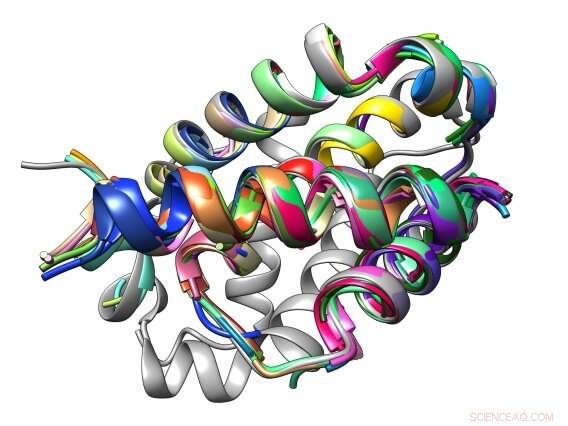
Bindningsgränssnittet mellan en peptid och dess Bcl-2-proteinmål är sammansatt av vanliga strukturella motiv som kallas TERM. Kredit:Sebastian Swanson och Avi Singer
Ett sätt att undersöka komplicerade biologiska system är att blockera deras komponenter från att interagera och se vad som händer. Denna metod gör det möjligt för forskare att bättre förstå cellulära processer och funktioner, förstärkning av vardagliga laboratorieexperiment, diagnostiska analyser, och terapeutiska insatser. Som ett resultat, reagenser som hindrar interaktioner mellan proteiner är mycket efterfrågade. Men innan forskare snabbt kan skapa sina egna anpassade molekyler som kan göra det, de måste först analysera det komplicerade förhållandet mellan sekvens och struktur.
Små molekyler kan lätt komma in i celler, men gränssnittet där två proteiner binder till varandra är ofta för stort eller saknar de små håligheter som krävs för att dessa molekyler ska kunna rikta in sig på. Antikroppar och nanokroppar binder till längre sträckor av protein, vilket gör dem bättre lämpade att hindra protein-proteininteraktioner, men deras stora storlek och komplexa struktur gör dem svåra att leverera och instabila i cytoplasman. Däremot korta sträckor av aminosyror, kända som peptider, är tillräckligt stora för att binda långa sträckor av protein samtidigt som de fortfarande är tillräckligt små för att komma in i celler.
Keating-labbet vid MIT Department of Biology arbetar hårt med att utveckla sätt att snabbt designa peptider som kan störa protein-protein-interaktioner som involverar Bcl-2-proteiner, som främjar cancertillväxt. Deras senaste tillvägagångssätt använder ett datorprogram som heter dTERMen, utvecklad av Keating lab alumn, Gevorg Grigoryan Ph.D. '07, för närvarande docent i datavetenskap och adjungerad docent i biologiska vetenskaper och kemi vid Dartmouth College. Forskare matar helt enkelt programmet med sina önskade strukturer, och det spottar ut aminosyrasekvenser för peptider som kan störa specifika protein-proteininteraktioner.
"Det är ett så enkelt sätt att använda, säger Keating, en MIT professor i biologi och senior författare på studien. "I teorin, du kan lägga in vilken struktur som helst och lösa en sekvens. I vår studie, programmet kom med nya sekvenskombinationer som inte liknar något som finns i naturen – det härledde ett helt unikt sätt att lösa problemet. Det är spännande att upptäcka nya territorier i sekvensuniversumet."
Tidigare postdoc Vincent Frappier och Justin Jenson Ph.D. '18 är medförfattare i studien, som finns i senaste numret av Strukturera .
Samma problem, annorlunda tillvägagångssätt
Jenson, för hans del, har tacklat utmaningen att designa peptider som binder till Bcl-2-proteiner med hjälp av tre distinkta tillvägagångssätt. Den dTERMen-baserade metoden, han säger, är den överlägset mest effektiva och generella han testat hittills.
Standardmetoder för att upptäcka peptidinhibitorer involverar ofta modellering av hela molekyler ner till fysiken och kemin bakom enskilda atomer och deras krafter. Andra metoder kräver tidskrävande skärmar för de bästa bindningskandidaterna. I båda fallen, processen är mödosam och framgångsfrekvensen är låg.
dTERMen, däremot kräver varken fysik eller experimentell screening, och utnyttjar vanliga enheter av kända proteinstrukturer, som alfaspiraler och betasträngar - kallade tertiära strukturella motiv eller "TERM" - som sammanställs i samlingar som Protein Data Bank. dTERMen extraherar dessa strukturella element från databanken och använder dem för att beräkna vilka aminosyrasekvenser som kan anta en struktur som kan binda till och avbryta specifika protein-proteininteraktioner. Det tar en enda dag att bygga modellen, och bara sekunder för att utvärdera tusen sekvenser eller designa en ny peptid.
"dTERMen tillåter oss att hitta sekvenser som sannolikt har de bindningsegenskaper vi letar efter, i en robust, effektiv, och allmänt sätt med hög framgångsgrad, " säger Jenson. "Tidigare tillvägagångssätt har tagit år. Men med hjälp av dTERMen, Vi gick från strukturer till validerade konstruktioner på några veckor."
Av de 17 peptiderna de byggde med de designade sekvenserna, 15 bundna med infödd-liknande affinitet, störa Bcl-2-protein-protein-interaktioner som är notoriskt svåra att rikta in sig på. I vissa fall, deras design var förvånansvärt selektiv och bundna till en enda Bcl-2 familjemedlem framför de andra. De designade sekvenserna avvek från kända sekvenser som finns i naturen, vilket kraftigt ökar antalet möjliga peptider.
"Denna metod tillåter en viss nivå av flexibilitet, " säger Frappier. "dTERMen är mer robusta mot strukturella förändringar, vilket gör att vi kan utforska nya typer av strukturer och diversifiera vår portfölj av potentiella bindande kandidater."
Undersöker sekvensuniversumet
Med tanke på de terapeutiska fördelarna med att hämma Bcl-2-funktionen och bromsa tumörtillväxt, Keating-labbet har redan börjat utöka sina designberäkningar till andra medlemmar av Bcl-2-familjen. De har för avsikt att så småningom utveckla nya proteiner som antar strukturer som aldrig tidigare setts.
"Vi har nu sett tillräckligt många exempel på olika lokala proteinstrukturer för att beräkningsmodeller av sekvens-struktursamband kan härledas direkt från strukturella data, snarare än att behöva återupptäckas varje gång från atomistiska interaktionsprinciper, säger Grigoryan, dTERMens skapare. "Det är oerhört spännande att en sådan strukturbaserad slutledning fungerar och är tillräckligt noggrann för att möjliggöra robust proteindesign. Det ger ett fundamentalt annorlunda verktyg för att hjälpa till att ta itu med strukturbiologins nyckelproblem - från proteindesign till strukturförutsägelse."
Frappier hoppas en dag kunna screena hela det mänskliga proteomet beräkningsmässigt, använda metoder som dTERMen för att generera kandidatbindande peptider. Jenson föreslår att användning av dTERMen i kombination med mer traditionella metoder för sekvensomdesign kan förstärka ett redan kraftfullt verktyg, ge forskare möjlighet att producera dessa riktade peptider. Helst han säger, att en dag utveckla peptider som binder och hämmar ditt favoritprotein kan vara lika enkelt som att köra ett datorprogram, eller lika rutin som att designa en DNA-primer.
Enligt Keating, även om den tiden fortfarande ligger i framtiden, "Vår studie är det första steget mot att visa denna kapacitet på ett problem av blygsam omfattning."
Den här historien återpubliceras med tillstånd av MIT News (web.mit.edu/newsoffice/), en populär webbplats som täcker nyheter om MIT-forskning, innovation och undervisning.