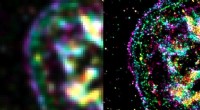
Ett exempel på sekundär strukturdetektering i kryo-EM-densitetskarta med Emap2Sec. En vänster är en EM-karta över archaeal 20S proteasom (EMDB ID:EMD-1733). Till höger detekteras sekundära strukturer av Emap2Sec. Punkter i magenta är positionerna för detekterade alfaspiraler; gula punkter detekteras betasträngar, och gröna punkter är för detekterade spolar (andra strukturer). Kredit:Purdue University bild/Daisuke Kihara
Kryoelektronmikroskopi är nu den mest populära metoden för att bestämma proteinstrukturer, som hjälper forskare att utveckla läkemedel mot olika sorters åkommor. Under de senaste decennierna, den har ersatt röntgenkristallografi eftersom den kan avbilda proteiner som inte lätt kan formas till stora kristaller. Den nya tekniken var så revolutionerande att den vann sina utvecklare 2017 års Nobelpris i kemi.
Slutprodukten av cryo-EM är en karta över tätheten av atomer i biologiska molekyler, men för att uppnå den detaljnivå som forskare behöver, de behöver göra ytterligare analyser. En ny studie i tidskriften Naturmetoder beskriver en teknik för att få lågupplösta kartor upp till par.
Vilken metod forskarna använder för att göra detta beror på vilken detaljnivå de börjar med. Kartor vid 2 till 3 ångström (Å, en längdenhet som används för att uttrycka storleken på atomer och molekyler) anses allmänt vara högupplösta. Dock, kartor av denna kvalitet är svåra att uppnå, och många produceras fortfarande vanligtvis i intervallet 4 till 10 Å. Av alla proteiner som deponerats till Elektronmikroskopdatabanken 2016-18, mer än 50 % löstes med medelupplösning.
"Om upplösningen är bättre än tre, då kan konventionella verktyg spåra aminosyraposition och bygga en karta över atompositioner. Men ofta kan cryo-EM inte ge dig en 3 Å-karta, sa Daisuke Kihara, professor i biologiska vetenskaper och datavetenskap vid Purdue University. "På kartor på 5 Å eller lägre, du kan vanligtvis inte se kedjeanslutningar alls."
Proteiner är faktiskt kedjor av aminosyror, och bindning mellan aminogrupper och karboxylgrupper skapar ibland vissa veckningsmönster. Dessa mönster, kända som alfaspiraler och betasträngar, bildar proteinets sekundära struktur.
På kartor från 5 till 8 Å, vissa fragment av den sekundära strukturen av proteiner är vanligtvis synliga, men att spåra hela kedjan skulle vara mycket svårt. Kiharas nya metod, känd som Emap2sec, avslöjar sekundära strukturer i kartor från 6 till 10 Å.
Emap2sec har ett djupt konvolutionellt neuralt nätverk i kärnan av sin algoritm. Dessa nätverk är djupinlärningssystem som främst används för att klassificera bilder, gruppera dem efter likhet och utför objektigenkänning. Det fungerar för identifiering av proteinstruktur i 3D-kartor eftersom metoden "konvolverar" lokala kartdensitetsegenskaper till bilder av en större region när informationen passerar genom lager av neurala nätverk. Den lokala förutsägelsen görs inom ramen för en stor del av kartan.
Identifierade sekundära strukturer i 3D-kartor hjälper forskare att tilldela kända strukturer av proteiner som redan har lösts in i kartan. Det betyder att de ibland har en utgångspunkt, eller åtminstone en aning om hur en del av strukturen ser ut. Emap2sec kan hjälpa forskare att få in sin bit i pusslet snabbare och enklare. Den identifierade strukturinformationen kan också vara till hjälp för att hitta fel i strukturmodellering.