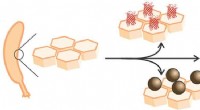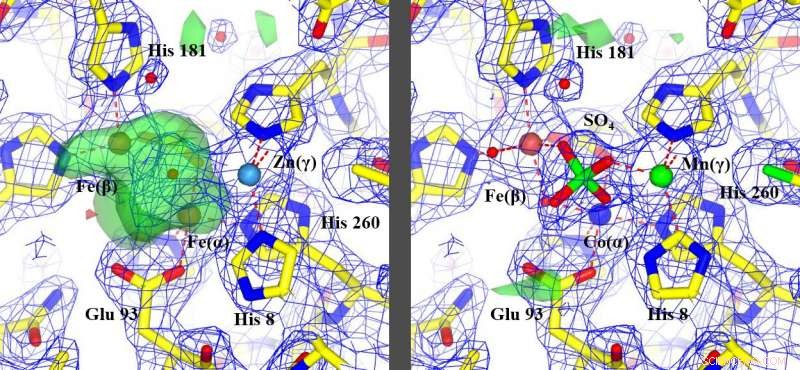
Den nya metoden, korrigera de felaktigt identifierade metallerna, tillåten omtolkning av oidentifierade egenskaper, markerad med grönt, (bild till vänster) för att identifiera hur proteinet fungerade, (bilden till höger). Kredit:Edward Snell
Proteiner som innehåller metall, kallas metalloproteiner, spelar viktiga roller inom biologi, reglerar olika vägar i kroppen, som ofta blir måltavlor för livräddande droger. Även om mängden metall i sådana proteiner vanligtvis är liten, det är avgörande för att bestämma funktionen hos dessa komplexa molekyler.
Forskare har länge vetat att metalloproteiner är avgörande för att förstå sjukdomar, som cancer, och för att utveckla nya läkemedel eftersom hämmare av metalloproteiner har använts för att behandla sjukdomar från cancer och HIV/AIDS till bakteriella infektioner och högt blodtryck. Men det har inte funnits en pålitlig analytisk metod för att bestämma identiteten och kvantiteten av metallatomer i metalloproteiner.
Nu, i en studie publicerad förra månaden i Journal of the American Chemical Society , ett internationellt team av forskare rapporterar att de har utvecklat ett sätt att entydigt identifiera och räkna metallatomer i proteiner på ett effektivt och rutinmässigt sätt. Använder det, teamet – som inkluderade forskare från universitetet i Buffalo, Hauptman-Woodward Medical Research Institute och andra – avslöjade ny information som fanns där, men tidigare gömd.
Kallas partikelinducerad emission av röntgenstrålar, eller PIXE, metoden utvecklades först på 1990-talet av Elspeth F. Garman från University of Oxford och Geoffrey W. Grime från University of Surrey Ion Beam Centre, båda författarna på den aktuella tidningen.
Det genombrott som rapporteras i den aktuella artikeln är utvecklingen av metoden till ett effektivt tillvägagångssätt med hög genomströmning och kombinationen med andra experimentella data för att identifiera typen och den exakta positionen för metallerna i proteinerna. Detta tillåter många olika typer av proteiner, ett stort antal som utgör livet som vi känner det, analyseras snabbt och effektivt, och ger ny information för en bättre strukturell förståelse.
Teamet tillämpade den nya metoden på 30 slumpmässigt utvalda metalloproteiner, som redan finns i det globala förrådet av proteinstrukturer som kallas Protein Data Bank. Det som hände därefter chockade dem.
"Resultatet var fantastiskt"
"Jag satt i Buffalo med min medarbetare från Oxford och när vi knackade siffrorna, vi båda insåg direkt att vi hade gjort en upptäckt, "mindes Edward Snell, Ph.D., en av motsvarande författare, som är VD och koncernchef för Hauptman-Woodward och professor vid Institutionen för materialdesign och innovation, ett gemensamt program för UB:s tekniska högskola och dess högskola för konst och vetenskap. "Vi gjorde siffrorna till en bild och dolda i data var en förklaring till hur denna molekylära maskin fungerade.
"Vi var först i världen att se vad som hade gömt sig där hela tiden. Resultatet var fantastiskt."
Resultaten visade att de metoder som tidigare använts för att bestämma några av dessa 30 slumpmässiga proteinstrukturer antingen hade felidentifierat metallatomen eller, i vissa fall, missade det helt.
"Enligt våra resultat, den nuvarande kunskapen om ungefär hälften av de prover vi studerade är felaktig, sa Snell.
Forskarna noterade att Protein Data Bank är en viktig resurs för forskare över hela världen. Under 2017, det gjordes i genomsnitt 1,86 miljoner nedladdningar per dag bara i USA. De konstaterar att ett enormt antal forskare använder strukturer från databanken utan kunskap om de potentiella grundläggande fel som kan finnas.
Och för närvarande, mer än 30 % av databanksmodellerna innehåller en metall.
Djupgående implikationer
"Att extrapolera från våra resultat där det fanns en felidentifierad metall i minst hälften av de studerade proverna tyder på att över 350, 000 modeller som laddas ner per dag kanske inte innehåller rätt metall, ", sade Snell. "Detta har djupgående konsekvenser för dem som använder modellerna. Om dessa modeller har fel, förståelsen för de miljontals människor som använder dem blir bristfällig."
Snell förklarade att en av svårigheterna med att studera metaller i proteiner är att de är mycket känsliga för röntgenstrålning, så experimentet i sig kan förändra det du ser. Men han noterade, en teknik som använder X-Ray Free Electron Lasers (XFELs), förhindrar detta eftersom experimenten vanligtvis är snabbare än någon förändring som kan inträffa.
Snell leder National Science Foundation BioXFEL Science and Technology Center, (Biology with X-ray Free Electron Lasers) ett konsortium av UB, Hauptman-Woodward och deras partners. Centret är dedikerat till att använda XFELs, som producerar otroligt intensiva röntgenstrålar i extremt korta pulser, och kan hjälpa till med en korrekt förståelse av dessa metaller i biologiska system.
Baserat på hans erfarenhet av Hauptman-Woodwards kristalliseringscentral med hög genomströmning, Snell samarbetade för att implementera PIXE-tekniken i en miljö med hög genomströmning. Han använde sin kunskap om röntgenegenskaper för att identifiera att ny strukturell information fanns i datan och tog sedan den kunskapen och förvandlade den till ett strukturellt resultat.
"I grund och botten, mina kollegor identifierade metallerna och vårt arbete i Buffalo visade dem var de skulle placeras, avslöjar den nya informationen som blev tillgänglig när metallen i modellen var korrekt, " han sa.