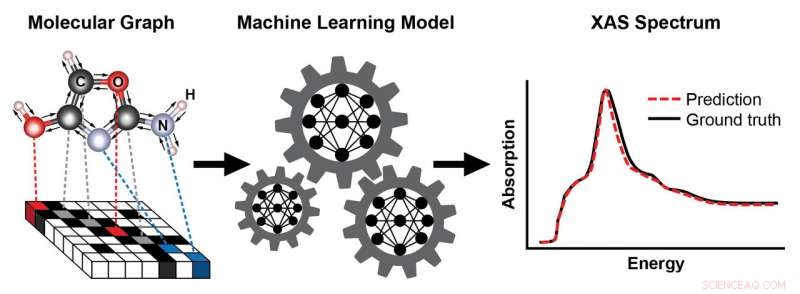
Ett schema som visar stegen för att träna en maskininlärningsmodell för att förutsäga ett röntgenabsorptionsspektrum (XAS) baserat på en molekyls kända struktur. Molekylens struktur representeras som en graf, med atomer som noder och kemiska bindningar som kanter. Denna representation fångar atomernas anslutningsmöjligheter – här, kol (C), syre (O), kväve (N), och väte (H) – och typen och längden på de kemiska bindningar som förbinder dem. Det resulterande XAS-spektrumet innehåller rik information om den lokala kemiska miljön för absorberande atomer, såsom deras symmetri och antalet närliggande atomer. Kredit:Brookhaven National Laboratory
Röntgenabsorptionsspektroskopi (XAS) är en populär karakteriseringsteknik för att undersöka den lokala atomstrukturen och elektroniska egenskaper hos material och molekyler. Eftersom atomer av varje element absorberar röntgenstrålar vid karakteristiska energier, XAS lämpar sig väl för att kartlägga den rumsliga fördelningen av element i ett prov. Vanligtvis, forskare utför XAS-experiment vid synkrotronljuskällor – som National Synchrotron Light Source II (NSLS-II) – eftersom de ger mycket ljus, inställbara röntgenstrålar. Genom att mäta absorbansen i ett prov vid varierande röntgenenergier, forskare kan skapa en plot som kallas ett röntgenabsorptionsspektrum.
"XAS är en nyckelfunktion för användare vid Brookhaven National Laboratorys NSLS-II och Center for Functional Nanomaterials (CFN), både US Department of Energy (DOE) Office of Science användarfaciliteter som är öppna för den vetenskapliga forskarvärlden, " sa Deyu Lu, en fysiker i CFN Theory and Computation Group. "Med rätt analysverktyg, XAS kan ge enorma insikter inom nanovetenskaplig forskning. Utvecklingen av sådana verktyg är central för vårt uppdrag som användaranläggning."
Klassificering av lokala kemiska miljöer
Olika områden av röntgenabsorptionsspektrat är känsliga för olika aspekter av materialegenskaperna i ett prov. Till exempel, röntgenabsorptionsnära-kantstrukturen (XANES) fokuserar på närkantsområdet av spektrumet, precis ovanför startenergin tillräcklig för att excitera en elektron från en atoms inre skal till ett tomt tillstånd. XANES kodar för rik information om den lokala kemiska miljön för absorberande atomer i ett prov – inklusive deras geometriska koordination, symmetri, och laddningstillstånd (antalet elektroner som erhålls eller förloras från kemisk bindning). Men att analysera spektraldata är mycket utmanande på grund av deras abstrakta natur.
"Till skillnad från en mikroskopbild av ett material där du direkt kan se egenskaper som kristallinitet eller defekter, XANES-spektra kodar information som kräver domänexpertis för att tolka, " förklarade Lu.
Standardtolkning av signaler i ett XANES-spektrum bygger på karakteristiska egenskaper som kallas "fingeravtryck, " som är konstruerade från mätningar på referensmaterial. denna fingeravtrycksmetod misslyckas när provet inte är en enkel kristall och relevanta referensmaterial inte lätt kan identifieras.
Storskaliga teoribaserade simuleringar från atomstrukturmodeller kan ge mycket användbara insikter för tolkningen av experimentella XANES-spektra; dock, dessa simuleringar är ofta beräkningsmässigt dyra och tidskrävande, och deras noggrannhetsnivå beror mycket på de valda teoretiska approximationerna och det system som studeras. Som ett resultat, Robust spektraltolkning är för närvarande flaskhalsen i XAS-studier. Vidare, realtidstolkning av XAS-spektra har dykt upp som en ny utmaning för studier av den dynamiska utvecklingen av material under driftsförhållanden och autonoma experiment. Behovet av robusta, effektiv spektraltolkning blir allt mer utbredd vid synkrotronljuskällor.
"Realtid, noggrann tolkning av röntgenspridning och spektroskopimätningar såsom röntgenabsorption, fluorescens, och diffraktion är en viktig förmåga för användare som bedriver forskning vid NSLS-II och andra synkrotronljusanläggningar, " sa Mehmet Topsakal, en vetenskaplig medarbetare i Materials for Energy Applications Group vid Brookhavens Nuclear Science and Technology Department som utvecklar avancerad dataanalys och maskininlärningstekniker för röntgenspektroskopi. "Varje år, tusentals forskare från hela världen kommer till NSLS-II för att undersöka egenskaperna hos olika material. En toppmodern spektralanalyspipeline skulle tillåta användare att få användbar feedback på sina prover medan experiment pågår och göra justeringar i farten för att vägleda experiment. Frågan är, hur kan vi göra spektraltolkning i realtid för att avslöja struktur-spektrumkorrelationer?"
Extrahera information med maskininlärning
Utnyttja big data och maskininlärning, Lu och Topsakal gav sig i kast med att svara på denna fråga tillsammans med beräkningsforskaren Shinjae Yoo från Brookhaven Labs Computational Science Initiative (CSI) och Columbia University Ph.D. kandidat och DOE Computational Science Graduate Fellow Matthew Carbone.
"DOE Computational Science Graduate Fellowship har gett mig en unik möjlighet att sträcka sig bortom min doktorsexamen i kemisk fysik vid Columbia för att utforska kraften i maskininlärningsalgoritmer, arbetar tillsammans med Brookhaven-forskare, ", sa Carbone. "Maskininlärning utnyttjar massiva datauppsättningar för att bygga mycket tydliga modeller som, en gång tränad, kan göra on-the-fly förutsägelser om nya data. Sådana modeller skulle kunna användas för att kringgå dyra kvantkemiberäkningar och stöd för karakterisering av operandomaterial."
Medlemmar i detta team och samarbetspartners har arbetat med spektrum-till-struktur- och struktur-till-spektrum-kartläggningar i flera år. Under 2017, de utvecklade maskininlärningsmodeller för att förutsäga det genomsnittliga koordinationstalet för metallnanopartiklar från XANES-spektra. Förra året, de skapade en XANES-databas för att lösa den lokala strukturen hos en amorf titanoxidbeläggning för fotokatalytiska tillämpningar. De byggde också en maskininlärningsmodell som kan förutsäga den lokala symmetrin hos absorbatoratomer från simulerade XANES-spektra av övergångsmetalloxider.
"När man utför spektraltolkning baserad på domänexpertis, vi tenderar att fokusera på specifika egenskaper konstruerade utifrån vår intuition, ", sa Lu. "Maskininlärning kan extrahera den information vi behöver på ett statistiskt framträdande sätt som eliminerar mänsklig fördom."
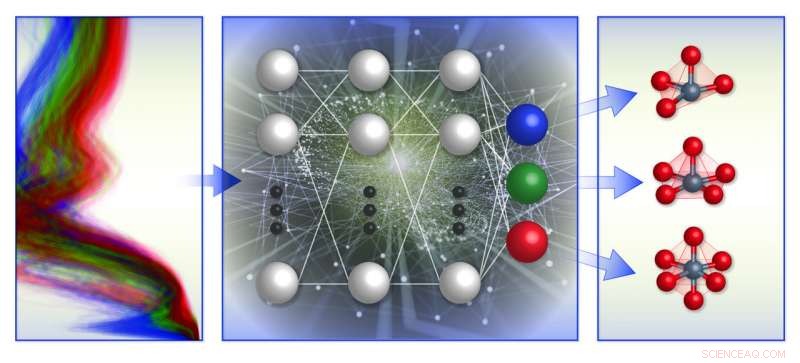
En schematisk illustration av teamets spektrumbaserade ramverk för klassificering av lokal kemisk miljö. De tränade maskininlärningsmodeller (mitten) med beräkningsdatabas för röntgenabsorptionsspektra (vänster) för att förutsäga den lokala geometrin kring positivt laddade övergångsmetalljoner (höger). Kredit:Brookhaven National Laboratory
Förutsäga röntgenabsorptionsspektra
Att bygga på sina tidigare framgångar, teamet tog sig an ett mer utmanande problem:träna en maskininlärningsmodell för att snabbt förutsäga spektra baserat på kända molekylära strukturer. En sådan modell skulle kringgå behovet av beräkningsdyra simuleringar, som inte är genomförbara under operandoexperiment, när forskare studerar material under driftsförhållanden. Trots växande maskininlärningsansträngningar för att förutsäga de kemiska egenskaperna hos material, direkta förutsägelser av de spektrala funktionerna hos verkliga material hade ännu inte uppnåtts.
"En teknisk svårighet är att bygga en optimal representation av molekylära strukturer som kan koda den inneboende symmetrin hos molekylerna som indatafunktioner för maskininlärningsmodellen, " sa Yoo.
Genom att anta en idé som nyligen föreslagits av forskare på Google, Topsakal och Carbone byggde en maskininlärningsmodell baserad på en grafrepresentation av molekyler som input, där atomer representeras som noder och kemiska bindningar som kanter.
"Datorer kan inte se molekyler som vi gör, ", sa Topsakal. "En graf är ett naturligt sätt att koda strukturen och anslutningsmöjligheterna hos en molekyl – fånga vilka atomer som är anslutna och typen och längden på de kemiska bindningarna som förbinder dem. Dessutom, denna representation är oföränderlig för transformationer såsom translationer och rotationer. Detta koncept är analogt med det inom bildigenkänning, där ett föremål som en katt eller hund i bakgrunden fortfarande kan klassificeras korrekt efter att bilden har transformerats."
För att träna modellen för en demonstration av principbevis, teamet använde en väletablerad databas (kallad QM9) som innehöll beräknad strukturell och kemisk information om 134, 000 små molekyler med upp till nio tunga atomer per atomtyp (kol, kväve, syre, och fluor). Från denna databas, de valde två träningsundergrupper – en undergrupp med molekyler som innehåller minst en syreatom, och en annan undergrupp med molekyler som innehåller minst en kväveatom - och beräknade deras motsvarande XANES-spektra. Sedan, de använde sina tränade modeller för att förutsäga XANES-spektra för syre- och kväveabsorptionskanter motsvarande excitationer av elektroner i det innersta skalet av respektive atom.
Maskininlärningsmodellen reproducerade nästan alla signifikanta absorptionstoppar och förutspådde topppositionerna (energier vid vilka toppar uppträder) och höjder (absorptionsintensiteter) med mycket hög noggrannhet. Modellen plockade också automatiskt upp domänens kunskap om att röntgenabsorptionsspektroskopi är känslig för funktionella grupper, eller grupper av atomer med liknande kemiska egenskaper och reaktivitet. Beroende på vilken funktionell grupp absorbatoratomen tillhör, olika egenskaper förekommer i spektrat.
"Vi är de första som visar att en maskininlärningsmodell kan användas för att exakt förutsäga fulla spektrala funktioner hos verkliga fysiska system direkt från deras strukturer, ", sa Topsakal. "Även om vi fokuserade på röntgenabsorptionsspektroskopi i vår studie, denna metod skulle kunna generaliseras för att förutsäga spektral information för andra populära tekniker, inklusive infraröd och gammastrålningsspektroskopi."
"När vi tränar maskininlärningsmodellen, vi behöver inte köra tidskrävande fysiska simuleringar, som tar minuter, timmar, eller till och med dagar, " sa Yoo. "Vi möjliggjorde inte bara realtidsspektraförutsägelse utan också den samtidiga genereringen av hundratals och tusentals spektra slutledningar genom att använda flera grafikbehandlingsenheter, eller GPU:er. Sådan teknik är nyckeln till att möjliggöra automatiserade strållinjekontroller och påskynda vetenskaplig upptäckt. I kombination med metoder för att ta prov på materialstrukturer, sådana modeller kan användas för att snabbt screena relevanta strukturer för att driva materialdesign och upptäckt."
Nästa, teamet skulle vilja kombinera koncept från deras modell som förutsäger lokal symmetri från XANES-spektra och denna nya modell som förutsäger XANES-spektra från molekylära strukturer. I sista hand, deras mål är att extrahera mer omfattande information om den lokala kemiska miljön eller till och med strukturen hos hela molekyler från experimentella mätningar.
"Verktyg för maskininlärning, som de för bild- och taligenkänning och drogupptäckt, är under snabb utveckling, ", sa Lu. "Nyckeln är att ta reda på hur man anpassar dessa verktyg på ett innovativt sätt för att tackla materialvetenskapliga problem."
"Vårt mål med att utveckla artificiell intelligens och maskininlärningsteknik är att lösa unika vetenskapliga utmaningar genom att både anta de senaste tekniska genombrotten inom dessa områden och komma med nya tillvägagångssätt som bidrar till respektive forskningsgemenskap, " lade Yoo till.