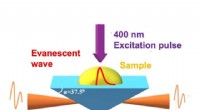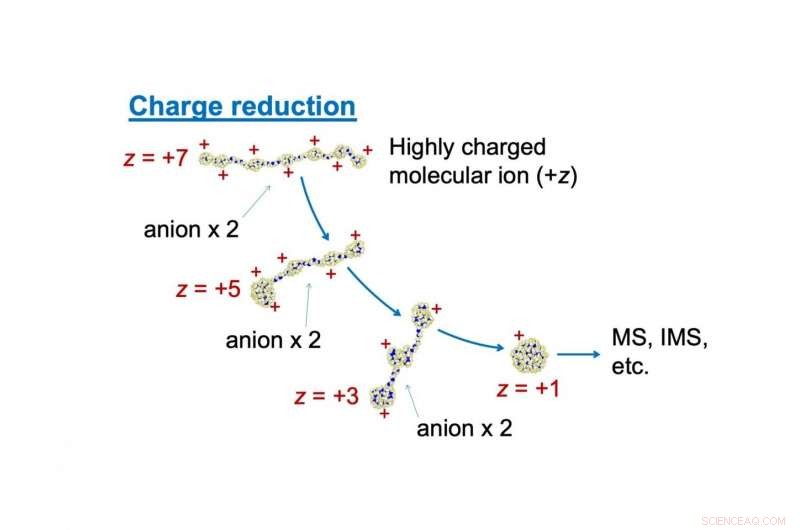
En skildring av laddningsreduceringsprocessen som används som en förbehandlingsprocess för masspektrometri, MS (eller jonmobilitetsspektrometri, IMS). För att öka spektrumets noggrannhet, överskott av laddningar på molekyljonen (PEG i denna figur) avlägsnas genom kollision med anjon i gas. Upphovsman:Kanazawa University
Masspektrometrar (MS) har blivit viktiga verktyg i kemi- och biologilaboratorier. Möjligheten att snabbt identifiera de kemiska komponenterna i ett prov gör att de kan delta i en mängd olika experiment, inklusive radiokolldatering, proteinanalys, och övervakning av läkemedelsmetabolism.
MS -instrument fungerar genom att ge analytmolekylerna en elektrisk laddning, och skjuter dem genom ett område av rymden med ett enhetligt elektriskt fält, som böjer sin bana till en cirkel. Cirkelns radie, som beror på förhållandet mellan molekylens massa och dess laddning, detekteras och jämförs med kända prover. Eftersom metoden bara kan mäta detta förhållande, inte själva massan, överskott kan leda till felaktiga eller tvetydiga resultat.
Nu, ett team av forskare under ledning av Kanazawa University använde en kraftfull molekylär dynamiksimulering för att bättre förstå effekten av överskottsladdningar på molekyler testade av en MS. De modellerade effekten av att tillsätta molekyler av motsatt laddning för att neutralisera överskottsladdning. I detta fall, den positiva laddningen på polyetylenglykol (PEG) kan reduceras genom kollision med negativt laddad NO 2 - joner.
Dock, detta kompliceras av det faktum att sannolikheten för att kollidera beror på laddningsbeloppet i första hand. "Laddade polymerer kan anta laddningsstatberoende strukturer på grund av elektrostatisk sträckning, "säger författaren Tomoya Tamadate. Till exempel, med liten överskottsavgift, PEG antar en kompakt form. Dock, när avgiften ökade, den ömsesidiga avstötningen mellan de positiva laddningarna får den att räta ut.
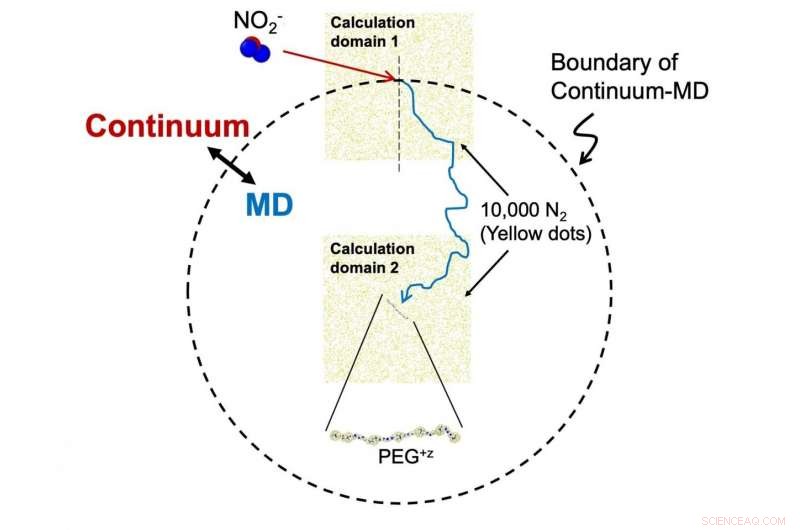
En kontur av utvecklad beräkningsmodell (kontinuum - molekylär dynamik simuleringshybridmetod). I denna modell, när avståndet mellan joner är tillräckligt långt, relativ rörelse beskrivs av diffusionsekvationer (kontinuum), under tiden inom ett visst avstånd (skrivet med streckad linje), molekylära dynamik (MD) simuleringar används för att beräkna banan. För att öka beräkningskostnaden. vi utför MD -simulering med att arrangera gasmolekyler endast runt måljoner. Upphovsman:Kanazawa University
För att påskynda beräkningarna, laget använde metoden "kontinuum approximation", som bara började simulera alla atomer i NO 2 - molekyl när den närmade sig tillräckligt nära PEG.
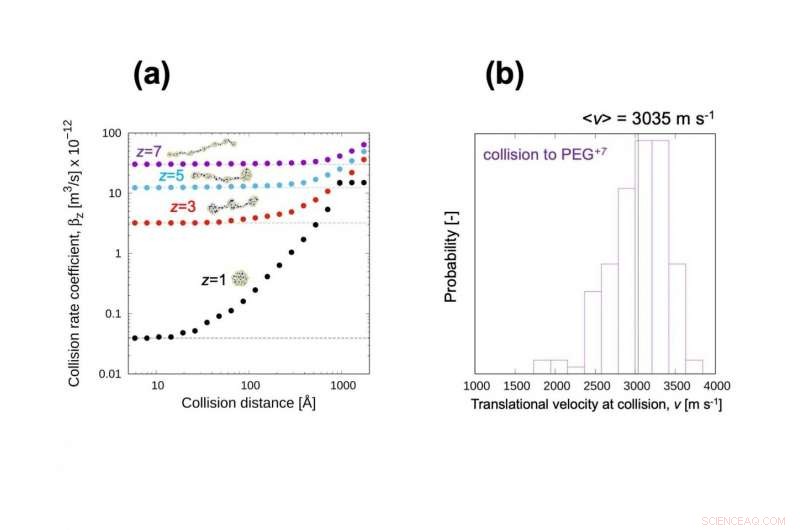
(a) Rekombination (kollision) koefficient för PEG -jon med olika antal laddningar. Denna kollisionshastighetskoefficient visade god överensstämmelse med experimentellt uppmätt laddningsreduceringshastighet. (b) Distributionshastighetsfördelning av NO 2 - jon vid kollision. Vi förväntar oss att rörelseenergin kan användas för att utvärdera möjligheten till kollisionsinducerad reaktion. Upphovsman:Kanazawa University
"Framgången för detta projekt visar att simuleringar av hybridkontinuum-molekylär dynamik kan användas mer allmänt för att studera kollisionsdrivna reaktionsmolekyler som kan anta olika konformationer, "säger författaren Takafumi Seto. Resultaten kan leda till effektivare metoder för att kontrollera överskottsladdning i provmolekyler, vilket möjliggör mer exakta resultat.