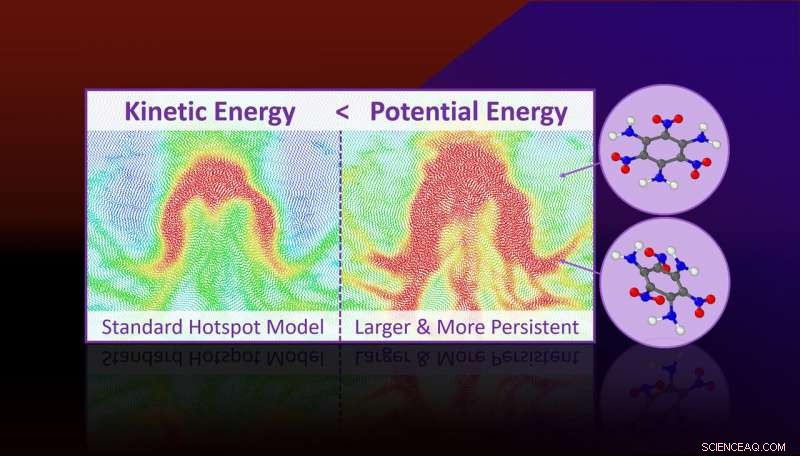
Molekylär dynamiksimuleringar förutspår att mer potentiell energi är lokaliserad i hotspots än vad deras kinetiska energi (eller temperatur) skulle antyda. Överskott av potentiell energi är knuten till ihållande ansträngda molekylära tillstånd som är förberedda för kemiska reaktioner och förklarar varför hotspots reagerar snabbare än bulken. Kredit:Lawrence Livermore National Laboratory
Forskning utförd på Lawrence Livermore National Laboratorys (LLNL) superdator Quartz belyser fynd gjorda av forskare som avslöjar en saknad aspekt av fysiken hos hotspots i TATB (1, 3, 5-trimamino-2, 4, 6-trinitrobensen) och andra sprängämnen.
Hotspots är lokaliserade områden med förhöjd temperatur som bildas från stötinducerad kollaps av mikrostrukturell porositet och är kända för att styra chockinitierings- och detonationsegenskaperna hos explosiva ämnen. Huvudkonceptet bakom hotspots är att lokala förhöjda temperaturer accelererar lokal kemi.
Forskningen presenteras i numret av den 11 mars av Journal of Physical Chemistry Letters och var ett samarbete mellan LLNL och Purdue University. Författare inkluderar Matthew Kroonblawd från LLNL och Brenden Hamilton, Chunyu Li och Alejandro Strachan från Purdue.
Arbetet belyser en försummad fysisk aspekt av de tidiga stadierna av hotspotbildning och evolution som ger en väg för att systematiskt förbättra multifysiska modeller för chockinitiering och detonation som används för att bedöma prestanda och säkerhet.
"Ett av de mest förbryllande resultaten från tidiga simuleringar av reaktiv molekylär dynamik är att hotspots som bildas vid kollapsade porer reagerar mycket snabbare än de av motsvarande storlek, temperatur och tryck i bulkmaterialet, ", sa Strachan. "Medan han kände igen, orsaken bakom dessa skillnader förstods inte. Vår studie löser denna fråga genom att vi finner att det explosiva materialet i en kollapsad por är fundamentalt annorlunda än bulken och att det är i ett högenergitillstånd förberedd för kemiska reaktioner."
Vikten av att förstå hotspots
TATB är ett okänsligt högexplosivt ämne som är kritiskt för landets kärnkraftslager och är utmanande att modellera i kontinuumskala. Tekniska modeller för explosiv säkerhet och detonationsprestanda bygger på fysikmodeller som fokuserar på bildandet och tillväxten av hotspots.
Kroonblawd förklarade att "multifysikmodeller på kontinuumnivå som används för att bedöma säkerhet och prestanda är mycket empiriska, vilket gör det svårt att skapa explosiva modeller som är överförbara till olika användningsförhållanden. Bristen på överförbara modeller gäller särskilt för okänsliga högexplosiva ämnen som TATB. Det är fortfarande inte möjligt att bygga en explosiv modell från första principer, vilket indikerar att nyckelaspekter saknas i vår förståelse av hotspotfysik och kemi."
Dessa modeller förlitar sig på noggranna behandlingar av kemisk reaktivitet och termisk transport; huruvida hotspots kommer att växa och sammansmälta till en detonationsvåg bestäms av en konkurrens mellan hastigheten för värmegenerering på grund av kemi och värmeförlust på grund av värmeledning.
Att identifiera orsaken bakom skillnader i hotspot-reaktionshastigheter ger en väg mot att formulera mer allmänna explosiva modeller som kommer att förbättra deras prediktiva noggrannhet och överförbarhet. Även om dessa modeller vanligtvis har fokuserat på temperatur som den huvudsakliga variabeln som styr kemin, resultaten tyder på att omarbetning av dessa modeller i termer av den potentiella energin kommer att ge en mer allmän behandling som kan särskilja de olika reaktiviteterna för olika materialtillstånd.
Genom simuleringar av all-atom molekylär dynamik, forskarna fann att hotspots inte bara är regioner med lokaliserad kinetisk energi (eller temperatur), men är också regioner med lokaliserad potentiell energi. Mängden potentiell energi är mycket större än mängden kinetisk energi och den koncentreras till molekylära moder som är relevanta för kemisk nedbrytning.
Den potentiella energilokaliseringen manifesterar sig på grund av spänningar på molekylnivå i plastiskt deformerade områden av materialet och detta kommer att leda till en mekanokemisk acceleration av reaktioner.
"Det viktigaste är att det inte finns något en-till-en-förhållande mellan kinetisk och potentiell energi i dessa system, därav, man kan inte sluta sig till lokala reaktionshastigheter från endast temperaturfältet, " sa Hamilton.
Teamet genomför storskaliga simuleringar
Arbetet, utförd av Materials Science Division personal i LLNL Energetic Materials Center (EMC) och Materials Engineering Department vid Purdue, fick stöd av LLNL:s Laboratory Directed Research and Development Strategic Initiative Program med Lara Leininger, EMC direktör, som huvudutredare. Arbetet innebar att köra storskaliga simuleringar av alla atomer på Livermore Computing-maskinen Quartz, och dessa simuleringar utfördes med hjälp av beräkningstid beviljad genom LLNL:s Computational Grand Challenge.
Att studera de långvariga avslappningsegenskaperna hos den kinetiska och potentiella energin i hotspots, teamet utvecklade en ny metod som heter Shock Trapping Internal Boundaries.
"Rent generellt, chocksimuleringar är begränsade i tid till när en chockvåg når nedströms simuleringsgränsen, som genererar tillståndsförändrande reflektionsvågor, " sa Hamilton. "I vår metod, vi kan isolera hotspot, eller någon region av intresse, förhindrar reflektioner från att interagera med det för att tillåta kontinuerliga studier av tidsutvecklingen."
Detta gjorde det möjligt för teamet att kvantifiera hastigheterna för avslappning av kinetisk och potentiell energi för att fastställa att den potentiella energin för hotspoten kvarstår efter att värmeledning försvinner den kinetiska energin.
Molekylär dynamiksimuleringar förutspår att mer potentiell energi är lokaliserad i hotspots än vad deras kinetiska energi (eller temperatur) skulle antyda. Överskott av potentiell energi är knuten till ihållande ansträngda molekylära tillstånd som är förberedda för kemiska reaktioner och förklarar varför hotspots reagerar snabbare än bulken.