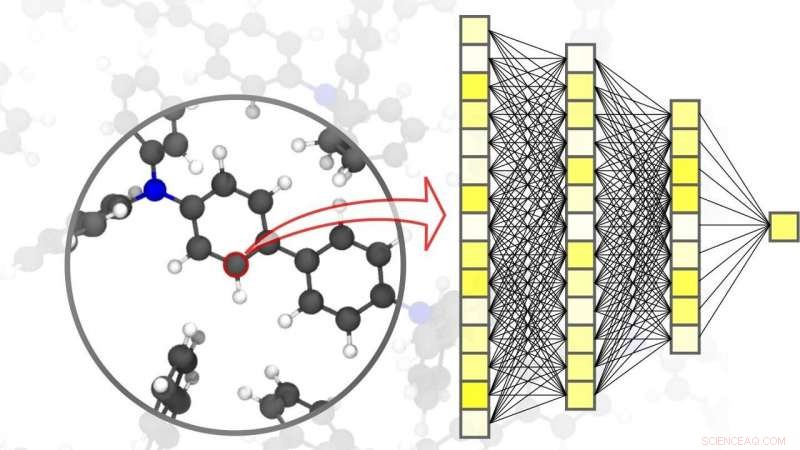
Neurala nätverk möjliggör exakta simuleringar inom materialvetenskap – ner till nivån för enskilda atomer. Kredit:Pascal Friedrich, UTRUSTNING
Forskning, utveckling, och produktion av nya material är mycket beroende av tillgången på snabba och samtidigt noggranna simuleringsmetoder. Maskininlärning, där artificiell intelligens (AI) självständigt förvärvar och tillämpar ny kunskap, kommer snart att göra det möjligt för forskare att utveckla komplexa materialsystem i en rent virtuell miljö. Hur fungerar detta, och vilka applikationer kommer att gynnas? I en artikel publicerad i Naturmaterial tidning, en forskare från Karlsruhe Institute of Technology (KIT) och hans kollegor från Göttingen och Toronto förklarar allt.
Digitalisering och virtualisering blir allt viktigare inom ett brett spektrum av vetenskapliga discipliner. En av dessa discipliner är materialvetenskap:forskning, utveckling, och produktion av nya material är mycket beroende av tillgången på snabba och samtidigt noggranna simuleringsmetoder. Detta, i tur och ordning, är fördelaktigt för ett brett utbud av olika applikationer – från effektiva energilagringssystem, sådana som är oumbärliga för användningen av förnybar energi, till nya mediciner, för vars utveckling krävs förståelse för komplexa biologiska processer. AI och maskininlärningsmetoder kan ta simuleringar inom materialvetenskap till nästa nivå. "Jämfört med konventionella simuleringsmetoder baserade på klassiska eller kvantmekaniska beräkningar, användningen av neurala nätverk speciellt anpassade för materialsimuleringar gör det möjligt för oss att uppnå en betydande hastighetsfördel, " förklarar fysikern och AI-experten professor Pascal Friederich, Chef för forskningsgruppen AiMat—Artificial Intelligence for Materials Sciences vid KIT:s Institute of Theoretical Informatics (ITI). "Med snabbare simuleringssystem, forskare kommer att kunna utveckla större och mer komplexa materialsystem i en rent virtuell miljö, och att förstå och optimera dem ner till atomär nivå."
Hög precision från atom till material
I en artikel publicerad i Naturmaterial , Pascal Friedrich, som också är biträdande gruppledare för divisionen Nanomaterials by Information-Guided Design vid KIT:s Institute of Nanotechnology (INT), presenterar, tillsammans med forskare från University of Göttingen och University of Toronto, en översikt över de grundläggande principerna för maskininlärning som används för simuleringar inom materialvetenskap. Detta inkluderar även datainsamlingsprocessen och aktiva inlärningsmetoder. Maskininlärningsalgoritmer möjliggör inte bara artificiell intelligens att bearbeta indata, men också för att hitta mönster och samband i stora datamängder, lära av dem, och göra autonoma förutsägelser och beslut. För simuleringar inom materialvetenskap, det är viktigt att uppnå hög noggrannhet över olika tids- och storleksskalor, allt från atom till material, samtidigt som beräkningskostnaderna begränsas. I deras artikel, forskarna diskuterar också olika aktuella tillämpningar, som små organiska molekyler och stora biomolekyler, strukturellt störd fast substans, flytande, och gasformiga material, såväl som komplexa kristallina system – till exempel, metallorganiska ramverk som kan användas för gaslagring eller för separation, för sensorer eller för katalysatorer.
Ännu högre hastighet med hybridmetoder
För att ytterligare utöka möjligheterna till materialsimuleringar i framtiden, forskarna från Karlsruhe, Göttingen, och Toronto föreslår utvecklingen av hybridmetoder:dessa kombinerar metoder för maskininlärning (ML) och molekylär mekanik (MM). MM-simuleringar använder så kallade kraftfält för att beräkna krafterna som verkar på varje enskild partikel och därmed förutsäga rörelser. Eftersom potentialerna för ML- och MM-metoderna är ganska lika, en tät integration med variabla övergångsområden är möjlig. Dessa hybridmetoder kan avsevärt påskynda simuleringen av stora biomolekyler eller enzymatiska reaktioner i framtiden, till exempel.