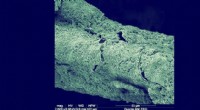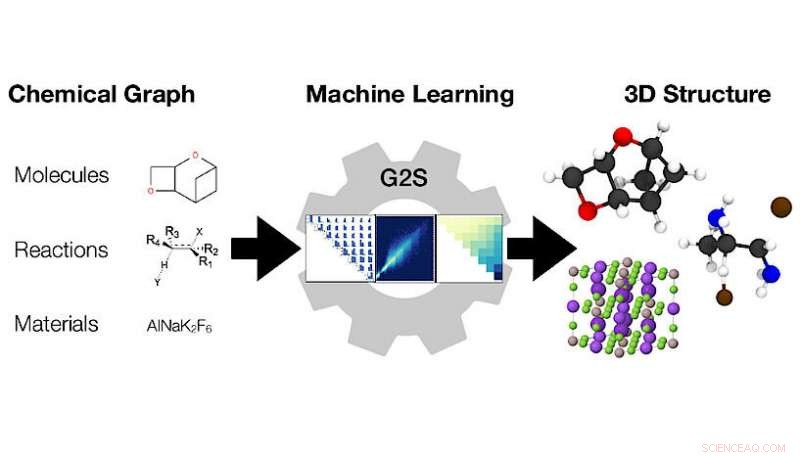
Maskininlärningsmodellen Graph2Structure använder grafer över kemiska föreningar (vänster) för att förutsäga deras 3D-koordinater (höger). Kredit:Dominik Lemm, Universitetet i Wien
3D-konfigurationer av atomer dikterar alla materialegenskaper. Kvantitativa förutsägelser av exakta jämviktsstrukturer, 3D-koordinater för alla atomer, från en kemisk graf, en representation av strukturformeln, är en utmanande och beräkningsmässigt dyr uppgift som är i början av praktiskt taget varje beräkningskemi-arbetsflöde. Forskare vid universitetet i Wien har nu utvecklat en ny maskininlärningsbaserad modell för att genväga dyra beräkningar för att direkt förutsäga strukturer från grafer. Den nya metoden för "maskininlärningsbaserade energifria strukturförutsägelser av molekyler, övergångstillstånd, och fasta ämnen" presenteras i senaste numret av Naturkommunikation .
Även om det ofta avbildas som stel, kemiska föreningar är flexibla tredimensionella föremål som består av atomer som kontinuerligt rör sig och oscillerar. Cyrus Levinthal noterade redan 1969 att den stora mängden frihetsgrader för kemiska föreningar formellt leder till ett katastrofalt stort antal möjliga konformationer långt upp till 10, 300 (Levinthals paradoxon). Inom experimentella observationer, dock, 3D-konfigurationer av atomer motsvarar väldefinierade minima för fri energi och dikterar därmed alla materialegenskaper. Paradigmet att struktur bestämmer funktion är nyckeln för att bestämma läkemedelsinteraktioner, optimera katalysatorer eller reaktioner, och materialupptäckt. Som en konsekvens, i de flesta beräkningskampanjer med hög genomströmning (en metod för snabba vetenskapliga experiment), endast de mest stabila konfigurationerna är eftertraktade. Beroende på graden av sofistikering inom de uppskattningar som görs vid uppskattning av materialens stabilitet, beräkningskostnaden kan variera från minuter till timmar eller till och med dagar för beräkning av en enskild struktur. Med tanke på det stora utrymmet för kemiska föreningar, utrymmet som befolkas av alla tänkbara föreningar (beräknas överstiga 1, 060) denna kompromiss mellan kostnad och kvalitet representerar en stor flaskhals på området.
Forskare vid universitetet i Wien under ledning av Anatole von Lilienfeld tacklade detta problem från ett annat perspektiv, utveckla en ny metod som utnyttjar data och är universellt tillämpbar på alla slags kemi. Deras nya metod, Graph2Structure, använder högkvalitativa kvantkemiska data för att träna maskininlärningsmodeller som kan förutsäga nya 3D-strukturer för molekylära grafer av osynliga föreningar. Denna direkta kartläggning av en molekylär graf till en specifik 3D-konfiguration gör det möjligt för modellen att effektivt kringgå alla former av energiminimering, vilket leder till en snabbare ökning på över en miljon jämfört med de konventionella metoderna. "Möjligheten att skapa högkvalitativa strukturer accelererar inte bara molekylär design med hög genomströmning, men påskyndar också det dagliga arbetsflödet, " säger huvudförfattaren till studien i Naturkommunikation Dominik Lemm. "Genererar tillförlitligt 3D-strukturer för även exotiska kemier, såsom öppna skalsystem eller övergångstillstånd, är en av de svåraste uppgifterna inom atomistisk simulering."
Ytterligare fynd tyder på att de genererade strukturerna direkt kan användas som en input till efterföljande utvärdering av maskininlärningsbaserade egenskapsprediktionsmodeller, därigenom koppla en molekylär graf till en strukturberoende egenskap på ett rigoröst och mer effektivt sätt.