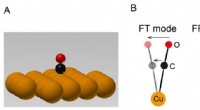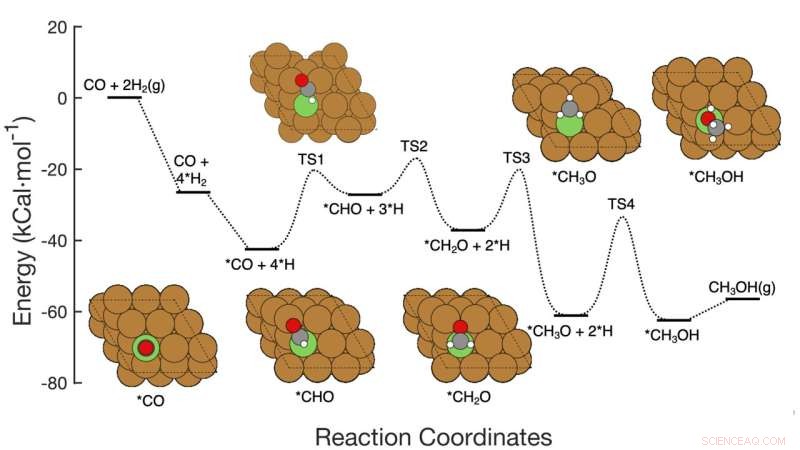
Denna grafik visar reaktionsvägen i sju steg för CO-hydrering till metanol över kopparbaserade katalysatorer, inklusive reaktanterna vid varje steg, schematiska atomarrangemang av mellanprodukterna och de energiaktiveringsbarriärer som krävs för att komma från steg till steg. Brookhaven Lab-teamet visade ett ramverk för maskininlärning som framgångsrikt identifierade vilka steg/kombinationer av steg som skulle justeras för att förbättra metanolproduktionen. Deras arbete kan hjälpa till att styra utformningen av nya katalysatorer för att uppnå det målet och ramverket kan tillämpas för att optimera andra reaktioner. Kredit:Brookhaven National Laboratory
Kemister vid det amerikanska energidepartementets Brookhaven National Laboratory har utvecklat ett nytt ramverk för maskininlärning (ML) som kan nollställa vilka steg i en flerstegs kemisk omvandling som bör justeras för att förbättra produktiviteten. Tillvägagångssättet kan hjälpa till att styra utformningen av katalysatorer - kemiska "dealmakers" som påskyndar reaktioner.
Teamet utvecklade metoden för att analysera omvandlingen av kolmonoxid (CO) till metanol med hjälp av en kopparbaserad katalysator. Reaktionen består av sju ganska enkla elementära steg.
"Vårt mål var att identifiera vilket elementärt steg i reaktionsnätverket eller vilken delmängd av steg som styr den katalytiska aktiviteten", säger Wenjie Liao, den första författaren på en artikel som beskriver metoden som just publicerats i tidskriften Catalysis Science &Technology . Liao är en doktorand vid Stony Brook University som har arbetat med forskare i gruppen Catalysis Reactivity and Structure (CRS) i Brookhaven Labs kemiavdelning.
Ping Liu, CRS-kemist som ledde arbetet, sa:"Vi använde den här reaktionen som ett exempel på vår ML-ramverksmetod, men du kan lägga in vilken reaktion som helst i det här ramverket i allmänhet."
Inriktning på aktiveringsenergier
Föreställ dig en kemisk reaktion i flera steg som en berg-och-dalbana med kullar av olika höjd. Höjden på varje kulle representerar den energi som behövs för att ta sig från ett steg till nästa. Katalysatorer sänker dessa "aktiveringsbarriärer" genom att göra det lättare för reaktanter att komma samman eller tillåta dem att göra det vid lägre temperaturer eller tryck. För att påskynda den övergripande reaktionen måste en katalysator inrikta sig på det eller de steg som har störst effekt.
Traditionellt skulle forskare som försöker förbättra en sådan reaktion beräkna hur man ändrar varje aktiveringsbarriär en i taget kan påverka den totala produktionshastigheten. Denna typ av analys kan identifiera vilket steg som var "hastighetsbegränsande" och vilka steg som avgör reaktionsselektiviteten – det vill säga om reaktanterna fortsätter till den önskade produkten eller längs en alternativ väg till en oönskad biprodukt.

Brookhaven Lab-kemist Ping Liu och Wenjie Liao, en doktorand vid Stony Brook University, utvecklade ett ramverk för maskininlärning för att identifiera vilka kemiska reaktionssteg som skulle kunna riktas mot för att förbättra reaktionsproduktiviteten. Kredit:Brookhaven National Laboratory
Men enligt Liu, "dessa uppskattningar blir väldigt grova med många fel för vissa grupper av katalysatorer. Det har verkligen skadat för katalysatordesign och screening, vilket är vad vi försöker göra", sa hon.
Det nya ramverket för maskininlärning är utformat för att förbättra dessa uppskattningar så att forskare bättre kan förutsäga hur katalysatorer kommer att påverka reaktionsmekanismer och kemisk produktion.
"Istället för att flytta en barriär i taget flyttar vi nu alla barriärer samtidigt. Och vi använder maskininlärning för att tolka den datamängden", sa Liao.
Detta tillvägagångssätt, sa teamet, ger mycket mer tillförlitliga resultat, inklusive om hur steg i en reaktion fungerar tillsammans.
"Under reaktionsförhållanden är dessa steg inte isolerade eller separerade från varandra, de är alla anslutna", sa Liu. "Om du bara gör ett steg i taget missar du mycket information - interaktionerna mellan de elementära stegen. Det är vad som har fångats i den här utvecklingen", sa hon.
Bygga modellen
Forskarna började med att bygga en datamängd för att träna sin maskininlärningsmodell. Datauppsättningen baserades på "density functional theory" (DFT) beräkningar av aktiveringsenergin som krävs för att transformera ett arrangemang av atomer till nästa genom reaktionens sju steg. Sedan körde forskarna datorbaserade simuleringar för att utforska vad som skulle hända om de ändrade alla sju aktiveringsbarriärerna samtidigt – några går upp, några går ner, några individuellt och några i par.
"Omfånget av data som vi inkluderade var baserat på tidigare erfarenheter av dessa reaktioner och detta katalytiska system, inom det intressanta variationsintervallet som sannolikt kommer att ge dig bättre prestanda," sa Liu.
Genom att simulera variationer i 28 "deskriptorer" – inklusive aktiveringsenergierna för de sju stegen plus stegpar som ändrades två åt gången – producerade teamet en omfattande datauppsättning med 500 datapunkter. Denna datauppsättning förutspådde hur alla dessa individuella tweaks och par av tweaks skulle påverka metanolproduktionen. Modellen fick sedan de 28 deskriptorerna efter deras betydelse för att driva metanolproduktionen.
"Vår modell "lärde sig" av data och identifierade sex nyckeldeskriptorer som den förutspår skulle ha störst inverkan på produktionen, säger Liao.
Efter att de viktiga deskriptorerna identifierats, tränade forskarna om ML-modellen med bara dessa sex "aktiva" deskriptorer. Denna förbättrade ML-modell kunde förutsäga katalytisk aktivitet enbart baserat på DFT-beräkningar för dessa sex parametrar.
"Istället för att du ska behöva beräkna hela 28 deskriptorerna kan du nu beräkna med endast de sex deskriptorerna och få de metanolomvandlingsfrekvenser du är intresserad av", sa Liu.
Teamet säger att de också kan använda modellen för att screena katalysatorer. Om de kan designa en katalysator som förbättrar värdet av de sex aktiva deskriptorerna, förutsäger modellen en maximal metanolproduktionshastighet.
Förstå mekanismer
När teamet jämförde förutsägelserna för deras modell med den experimentella prestandan hos deras katalysator - och prestandan hos legeringar av olika metaller med koppar - stämde förutsägelserna överens med de experimentella resultaten. Jämförelser av ML-metoden med den tidigare metoden som användes för att förutsäga legeringars prestanda visade att ML-metoden var mycket överlägsen.
Data avslöjade också en hel del detaljer om hur förändringar i energibarriärer kan påverka reaktionsmekanismen. Av särskilt intresse – och betydelse – var hur olika steg i reaktionen samverkar. Till exempel visade data att i vissa fall skulle en sänkning av energibarriären i det hastighetsbegränsande steget inte i sig förbättra metanolproduktionen. Men att justera energibarriären för ett steg tidigare i reaktionsnätverket, samtidigt som aktiveringsenergin för det hastighetsbegränsande steget hålls inom ett idealiskt område, skulle öka metanolproduktionen.
"Vår metod ger oss detaljerad information som vi kanske kan använda för att designa en katalysator som koordinerar interaktionen mellan dessa två steg väl," sa Liu.
Men Liu är mest exalterad över potentialen för att tillämpa sådana datadrivna ML-ramverk på mer komplicerade reaktioner.
"Vi använde metanolreaktionen för att demonstrera vår metod. Men hur den genererar databasen och hur vi tränar ML-modellen och hur vi interpolerar rollen för varje deskriptors funktion för att bestämma den totala vikten i termer av deras betydelse - det kan vara lätt appliceras på andra reaktioner", sa hon. + Utforska vidare