Maskininlärning (ML) möjliggör noggrann och effektiv beräkning av grundläggande elektroniska egenskaper hos binära och ternära oxidytor, vilket visades av forskare från Tokyo Tech. Deras ML-baserade modell skulle kunna utvidgas till andra föreningar och egenskaper. Resultaten, publicerade i Journal of the American Chemical Society , skulle kunna hjälpa till vid screening av ytegenskaper hos material såväl som vid utveckling av funktionella material.
Design och utveckling av nya material med överlägsna egenskaper kräver en omfattande analys av deras atomära och elektroniska strukturer.
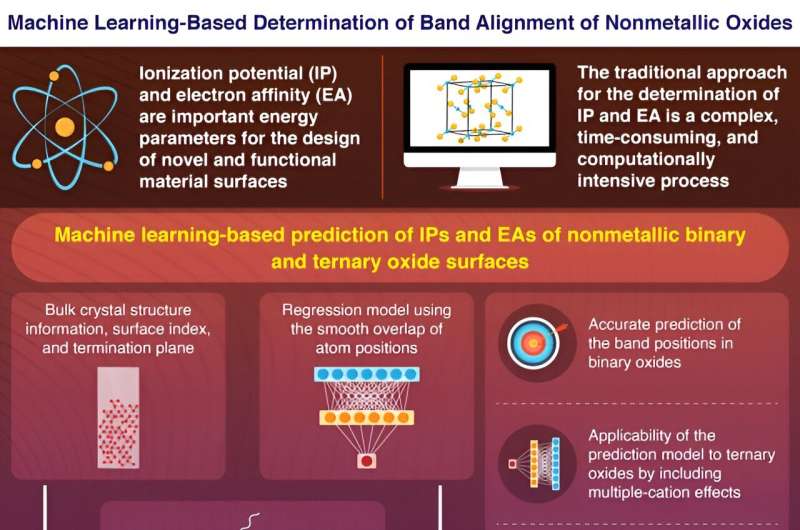
Elektronenergiparametrar som joniseringspotential (IP), energin som behövs för att ta bort en elektron från valensbandets maximum och elektronaffinitet (EA), mängden energi som frigörs vid anslutning av en elektron till ledningsbandets minimum, avslöjar viktiga information om den elektroniska bandstrukturen för ytor av halvledare, isolatorer och dielektrikum.
Den exakta uppskattningen av IP och EA i sådana icke-metalliska material kan indikera deras tillämplighet för användning som funktionella ytor och gränssnitt i ljuskänslig utrustning och optoelektroniska enheter.
Dessutom beror IP:er och EA:er avsevärt på ytstrukturerna, vilket ger ytterligare en dimension till det komplexa förfarandet för deras kvantifiering. Traditionell beräkning av IP:er och EA:er innebär användning av noggranna beräkningar av första principerna, där bulk- och ytsystem kvantifieras separat. Denna tidskrävande process förhindrar kvantifiering av IP:er och EA:er för många ytor, vilket kräver användning av beräkningseffektiva metoder.
För att ta itu med de omfattande problem som påverkar kvantifieringen av IP:er och EA:er av icke-metalliska fasta ämnen, har ett team av vetenskapsmän från Tokyo Institute of Technology (Tokyo Tech), ledd av professor Fumiyasu Oba, riktat sitt fokus mot ML.
Prof. Oba säger, "Under de senaste åren har ML fått mycket uppmärksamhet inom materialvetenskaplig forskning. Möjligheten att virtuellt screena material baserat på ML-teknologi är ett mycket effektivt sätt att utforska nya material med överlägsna egenskaper. Dessutom är förmågan att träna stora datamängder med hjälp av noggranna teoretiska beräkningar möjliggör en framgångsrik förutsägelse av viktiga ytegenskaper och deras funktionella implikationer."
Forskarna använde ett artificiellt neuralt nätverk för att utveckla en regressionsmodell, som inkluderade smidig överlappning av atompositioner (SOAP) som numerisk indata. Deras modell förutspådde noggrant och effektivt IP och EA för binära oxidytor genom att använda informationen om bulkkristallstrukturer och yttermineringsplan.
Dessutom kan den ML-baserade förutsägelsemodellen "överföra inlärning", ett scenario där en modell utvecklad för ett visst syfte kan göras för att införliva nyare datauppsättningar och återansöka för ytterligare uppgifter. Forskarna inkluderade effekterna av flera katjoner i sin modell genom att utveckla "lärbara" SOAP:er och förutspådde IP:er och EA för ternära oxider med hjälp av transfer learning.
Prof. Oba avslutar, "Vår modell är inte begränsad till förutsägelse av ytegenskaper hos oxider utan kan utvidgas till att studera andra föreningar och deras egenskaper."
Mer information: Shin Kiyohara et al, Band Alignment of Oxides by Learnable Structural-Descriptor-Aided Neural Network and Transfer Learning, Journal of the American Chemical Society (2024). DOI:10.1021/jacs.3c13574
Journalinformation: Tidskrift för American Chemical Society
Tillhandahålls av Tokyo Institute of Technology