Ett team av beräkningsforskare vid Department of Energy's Oak Ridge National Laboratory har genererat och släppt datauppsättningar av oöverträffad skala som tillhandahåller de ultravioletta synliga spektrala egenskaperna hos över 10 miljoner organiska molekyler. Att förstå hur en molekyl interagerar med ljus är avgörande för att avslöja dess elektroniska och optiska egenskaper, som i sin tur har potentiella fotoaktiva tillämpningar i produkter som solceller eller medicinska bildsystem.
Med hjälp av högpresterande beräkningsresurser vid Oak Ridge Leadership Computing Facility, körde ORNL-teamet kvantkemiberäkningar för att generera de enorma datamängderna. För var och en av dessa organiska molekyler körde teamet atomistiska materialmodelleringsberäkningar med olika uppskattningar för att beräkna olika intressanta egenskaper i exciterat tillstånd. Teamets resultat publicerades i Scientific Data .
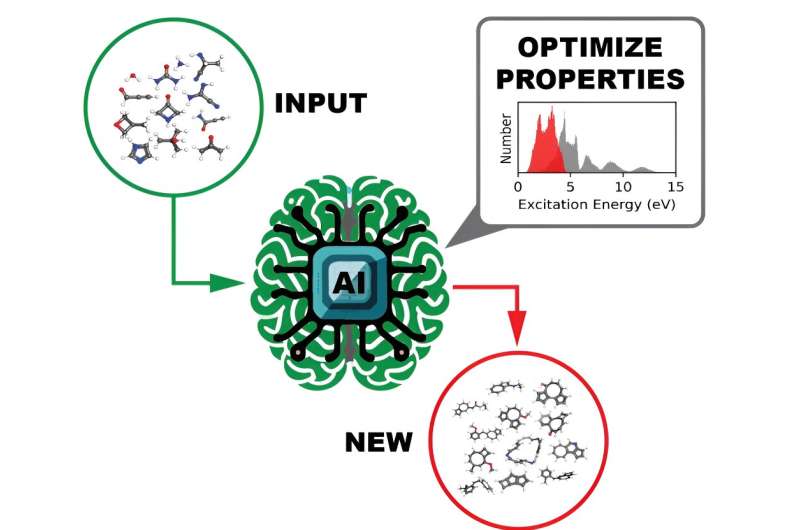
Den ultimata avsedda användningen för datauppsättningarna med öppen källkod är att träna en modell för djupinlärning för att identifiera molekyler med skräddarsydda optoelektroniska och fotoreaktivitetsegenskaper, ett tillvägagångssätt som är mycket snabbare och lättare att genomföra än nuvarande metoder.
"Användningen av DL-modeller för molekylär design är avgörande eftersom det kemiska utrymmet som måste utforskas för att söka efter dessa molekyler är extremt stort", säger huvudförfattaren Massimiliano Lupo Pasini, en dataforskare vid ORNL:s Computational Sciences and Engineering Division.
"Både experiment och befintliga beräkningar av första principerna, som är baserade på de fysiska lagarna som bestämmer hur materia och energi interagerar på subatomär nivå, är helt enkelt oöverkomliga av olika anledningar. Experiment är arbetsintensiva och beräkningar av första principerna kan lätt slå sönder superdatorer Men DL-modeller ger mycket lovande verktyg för att övervinna dessa hinder," sa Lupo Pasini.
Projektet kom igång när Stephan Irle, ledare för ORNL:s Computational Chemistry and Nanomaterials Sciences-grupp, identifierade de ultraviolett-synliga spektrumen av molekyler som en användbar egenskap att förutsäga med DL-modeller.
Att bygga en DL-modell som är tillräckligt komplex för att identifiera önskvärda molekylära egenskaper kräver att den tränas med enorma mängder data som utforskar alla olika regioner i det kemiska rummet. Ju mer data som samlas in, desto mer kan DL-modellen som tränas på den uppnå den nödvändiga robustheten och generaliserbarheten för att fungera effektivt. Men att samla in så stora mängder vetenskaplig data för skalbar DL kan ge problem med dataflödet, särskilt vid anläggningar med flera användare som OLCF, en DOE Office of Science-användaranläggning belägen på ORNL.
"En utmaning som uppstår när man genererar stora datamängder är att antalet filer som ska hanteras ökar drastiskt. Om den inte hanteras korrekt kan en så stor datamängd äventyra funktionen hos det parallella filsystemet, vilket är en viktig del av staten. -of-the-art HPC-anläggningar," sa Lupo Pasini.
För att möta denna utmaning, samarbetade Lupo Pasini med ORNL-datavetaren Kshitij Mehta för att utveckla en skalbar arbetsflödesprogramvara som säkerställer att filerna som genereras av kvantmekanikkoden hanteras korrekt utan att stressa filsystemet, såsom OLCF:s Orion, som är en delad resurs som hanterar inmatning, utmatning och lagring av data på superdatorsystem.
Som ett proof-of-concept-test genererade teamet GDB-9-Ex-datauppsättningen med 96 766 molekyler som består av kol, kväve, syre och fluor, med högst nio icke-väteatomer. Det visade att det designade arbetsflödet är effektivt och att DL-träningen exakt förutsäger positionen och intensiteten för de mest relevanta topparna i det ultraviolett-synliga spektrumet.
Från den första framgången ökade teamet sin volym med ORNL_AISD-Ex-datauppsättningen, som innehåller 10 502 917 molekyler bestående av kol, kväve, syre, fluor och svavel, med högst 71 icke-väteatomer. Pilsun Yoo, en postdoktor i Irles grupp, utvecklade verktyg för att analysera de resulterande datamängderna.
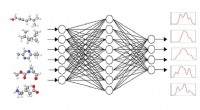
Det ultraviolett-synliga spektrumet, som beskriver en molekyls excitationslägen, beräknades för var och en av de mer än 10 miljoner molekylerna. Denna information avslöjar vilken ljusfrekvens som krävs för att rikta in sig på en molekyl och bryta isär vissa bindningar av den kemiska föreningen.
En annan egenskap av intresse som beräknades för varje molekyl var HOMO-LUMO-gapet - energigapet mellan den högsta ockuperade molekylära orbitalen och den lägsta lediga molekylorbitalen - som på ett tillförlitligt sätt mäter molekylens stabilitet. Med denna information kan en DL-modell effektivt sålla igenom data för att identifiera lovande molekyler för olika framtida användningar.
Faktum är att Lupo Pasini och hans team på ORNL, inklusive beräkningsforskaren i maskininlärning Pei Zhang och HPC-dataforskaren Jong Youl Choi, utvecklar just en sådan DL-modell:HydraGNN.
"HydraGNN-arkitekturen tar in den atomära strukturen, omvandlar den till en graf och sedan försöker den förutsäga som en utdata vad koden för första principerna skulle producera. Det är en surrogatmodell för dyra första principberäkningar," sa Lupo Pasini.
Resultaten från HydraGNN:s utbildning om datamängderna och dess molekylära upptäckter kommer att beskrivas i ett kommande dokument.
Mer information: Massimiliano Lupo Pasini et al, Två datauppsättningar av exciterade tillstånd för kvantkemiska UV-vis spektra av organiska molekyler, Scientific Data (2023). DOI:10.1038/s41597-023-02408-4
Journalinformation: Vetenskapliga data
Tillhandahålls av Oak Ridge National Laboratory