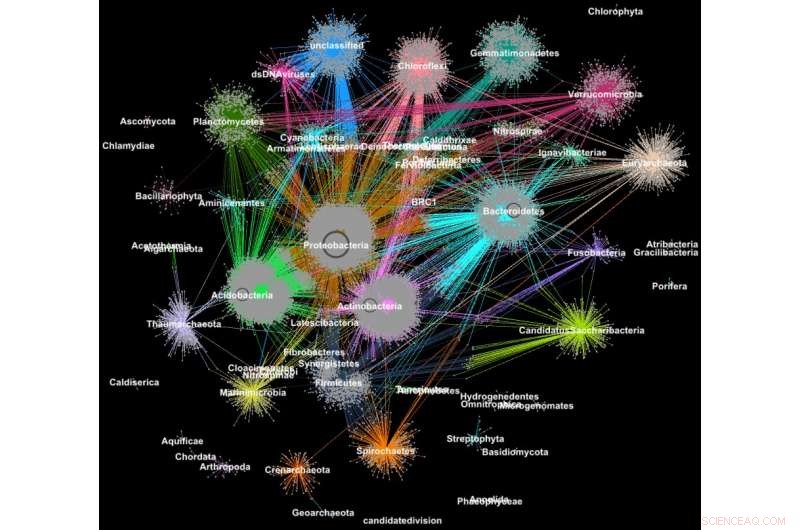
Proteiner från metagenomer grupperade i familjer enligt deras taxonomiska klassificering. Kredit:Georgios Pavlopoulos och Nikos Kyrpides, JGI/Berkeley Lab
Visste du att verktygen som används för att analysera relationer mellan användare av sociala nätverk eller ranka webbsidor också kan vara oerhört värdefulla för att förstå stora vetenskapliga data? På ett socialt nätverk som Facebook, varje användare (person eller organisation) representeras som en nod och kopplingarna (relationer och interaktioner) mellan dem kallas kanter. Genom att analysera dessa samband, forskare kan lära sig mycket om varje användare – intressen, hobbyer, köpvanor, vänner, etc.
I biologi, liknande graf-klustringsalgoritmer kan användas för att förstå de proteiner som utför de flesta av livets funktioner. Det uppskattas att människokroppen ensam innehåller cirka 100, 000 olika proteintyper, och nästan alla biologiska uppgifter – från matsmältning till immunitet – uppstår när dessa mikroorganismer interagerar med varandra. En bättre förståelse för dessa nätverk kan hjälpa forskare att fastställa effektiviteten av ett läkemedel eller identifiera potentiella behandlingar för en mängd olika sjukdomar.
I dag, avancerad teknik med hög genomströmning tillåter forskare att fånga hundratals miljoner proteiner, gener och andra cellulära komponenter på en gång och i en rad miljöförhållanden. Klustringsalgoritmer tillämpas sedan på dessa datauppsättningar för att identifiera mönster och samband som kan peka på strukturella och funktionella likheter. Även om dessa tekniker har använts i stor utsträckning i mer än ett decennium, de kan inte hålla jämna steg med strömmen av biologisk data som genereras av nästa generations sequencers och mikroarrayer. Faktiskt, mycket få befintliga algoritmer kan klustera ett biologiskt nätverk som innehåller miljontals noder (proteiner) och kanter (anslutningar).
Det är därför ett team av forskare från Department of Energys (DOE:s) Lawrence Berkeley National Laboratory (Berkeley Lab) och Joint Genome Institute (JGI) tog en av de mest populära klustringsmetoderna inom modern biologi – Markov Clustering (MCL) algoritmen – och ändrade den för att köras snabbt, effektivt och i stor skala på superdatorer med distribuerat minne. I ett testfall, deras högpresterande algoritm – kallad HipMCL – uppnådde en tidigare omöjlig bedrift:att samla ett stort biologiskt nätverk innehållande cirka 70 miljoner noder och 68 miljarder kanter på ett par timmar, använder cirka 140, 000 processorkärnor på National Energy Research Scientific Computing Centers (NERSC) Cori superdator. En artikel som beskriver detta arbete publicerades nyligen i tidskriften Nukleinsyraforskning .
"Den verkliga fördelen med HipMCL är dess förmåga att klustera massiva biologiska nätverk som var omöjliga att klustera med den befintliga MCL-mjukvaran, vilket gör det möjligt för oss att identifiera och karakterisera det nya funktionella utrymmet som finns i de mikrobiella samhällena, säger Nikos Kyrpides, som leder JGI:s Microbiome Data Science-satsningar och Prokaryote Super Program och är medförfattare på tidningen. "Dessutom kan vi göra det utan att offra något av den ursprungliga metodens känslighet eller noggrannhet, vilket alltid är den största utmaningen i den här typen av skalningsinsatser."
"När vår data växer, det blir ännu viktigare att vi flyttar våra verktyg till högpresterande datormiljöer, " tillägger han. "Om du skulle fråga mig hur stort är proteinutrymmet? Sanningen är den, vi vet inte riktigt eftersom vi hittills inte hade beräkningsverktygen för att effektivt samla alla våra genomiska data och undersöka den funktionella mörka materien."
Förutom framsteg inom datainsamlingsteknik, forskare väljer alltmer att dela sina data i gemenskapsdatabaser som systemet Integrated Microbial Genomes &Microbiomes (IMG/M), som utvecklades genom ett decennier gammalt samarbete mellan forskare vid JGI och Berkeley Labs Computational Research Division (CRD). Men genom att tillåta användare att göra jämförande analyser och utforska de funktionella kapaciteterna hos mikrobiella samhällen baserat på deras metagenomiska sekvens, gemenskapsverktyg som IMG/M bidrar också till dataexplosionen inom tekniken.
Hur slumpmässiga promenader leder till datorflaskhalsar
För att få grepp om denna ström av data, forskare förlitar sig på klusteranalys, eller klustring. Detta är i huvudsak uppgiften att gruppera objekt så att objekt i samma grupp (kluster) är mer lika än de i andra kluster. I mer än ett decennium, Beräkningsbiologer har gynnat MCL för klustring av proteiner genom likheter och interaktioner.
"En av anledningarna till att MCL har varit populärt bland beräkningsbiologer är att det är relativt parameterfritt; användare behöver inte ställa in massor av parametrar för att få korrekta resultat och det är anmärkningsvärt stabilt mot små förändringar i data. Detta är viktigt eftersom du kanske måste omdefiniera en likhet mellan datapunkter eller så kanske du måste korrigera för ett litet mätfel i dina data. I dessa fall du vill inte att dina ändringar ska ändra analysen från 10 kluster till 1, 000 kluster, " säger Aydin Buluç, en CRD-forskare och en av artikelns medförfattare.
Men, han lägger till, beräkningsbiologiska samfundet stöter på en datorflaskhals eftersom verktyget mestadels körs på en enda datornod, är beräkningsmässigt dyrt att köra och har ett stort minnesfotavtryck – vilket alla begränsar mängden data som denna algoritm kan klustera.
Ett av de mest beräknings- och minnesintensiva stegen i denna analys är en process som kallas random walk. Denna teknik kvantifierar styrkan hos en koppling mellan noder, vilket är användbart för att klassificera och förutsäga länkar i ett nätverk. I fallet med en internetsökning, detta kan hjälpa dig att hitta ett billigt hotellrum i San Francisco för spring break och till och med berätta när det är bäst att boka det. I biologi, ett sådant verktyg kan hjälpa dig att identifiera proteiner som hjälper din kropp att bekämpa ett influensavirus.
Givet en godtycklig graf eller nätverk, det är svårt att veta det mest effektiva sättet att besöka alla noder och länkar. En slumpmässig promenad får en känsla av fotavtrycket genom att utforska hela grafen slumpmässigt; den börjar vid en nod och rör sig godtyckligt längs en kant till en angränsande nod. Denna process fortsätter tills alla noder i grafnätverket har nåtts. Eftersom det finns många olika sätt att resa mellan noder i ett nätverk, detta steg upprepas flera gånger. Algoritmer som MCL kommer att fortsätta köra denna slumpmässiga gångprocess tills det inte längre finns en signifikant skillnad mellan iterationerna.
I vilket nätverk som helst, du kanske har en nod som är ansluten till hundratals noder och en annan nod med bara en anslutning. De slumpmässiga promenaderna kommer att fånga de mycket anslutna noderna eftersom en annan väg kommer att upptäckas varje gång processen körs. Med denna information, Algoritmen kan med viss säkerhet förutsäga hur en nod i nätverket är ansluten till en annan. Mellan varje slumpmässig promenad, Algoritmen markerar sin förutsägelse för varje nod på grafen i en kolumn i en Markov-matris – ungefär som en huvudbok – och slutliga kluster avslöjas i slutet. Det låter enkelt nog, men för proteinnätverk med miljontals noder och miljarder kanter, detta kan bli ett extremt beräknings- och minnesintensivt problem. Med HipMCL, Berkeley Labs datavetare använde avancerade matematiska verktyg för att övervinna dessa begränsningar.
"Vi har framför allt hållit MCL-ryggraden intakt, gör HipMCL till en massivt parallell implementering av den ursprungliga MCL-algoritmen, säger Ariful Azad, en datavetare i CRD och huvudförfattare till artikeln.
Även om det har gjorts tidigare försök att parallellisera MCL-algoritmen för att köras på en enda GPU, verktyget kunde fortfarande bara gruppera relativt små nätverk på grund av minnesbegränsningar på en GPU, Azad anteckningar.
"Med HipMCL omarbetar vi i huvudsak MCL-algoritmerna för att fungera effektivt, parallellt på tusentals processorer, och ställ in den för att dra nytta av det samlade minnet som är tillgängligt i alla beräkningsnoder, " tillägger han. "Den oöverträffade skalbarheten hos HipMCL kommer från dess användning av toppmoderna algoritmer för sparsam matrismanipulation."
Enligt Buluç, att utföra en slumpmässig promenad samtidigt från många noder i grafen beräknas bäst med gles-matrismatrismultiplikation, vilket är en av de mest grundläggande operationerna i den nyligen släppta GraphBLAS-standarden. Buluç och Azad utvecklade några av de mest skalbara parallella algoritmerna för GraphBLAS:s glesa matrismatrismultiplikation och modifierade en av deras toppmoderna algoritmer för HipMCL.
"Kruxet här var att hitta den rätta balansen mellan parallellism och minneskonsumtion. HipMCL extraherar dynamiskt så mycket parallellism som möjligt givet det tillgängliga minnet som tilldelats den, säger Buluç.
HipMCL:Clustering i skala
Förutom de matematiska innovationerna, en annan fördel med HipMCL är dess förmåga att köras sömlöst på alla system – inklusive bärbara datorer, arbetsstationer och stora superdatorer. Forskarna uppnådde detta genom att utveckla sina verktyg i C++ och använda standard MPI- och OpenMP-bibliotek.
"Vi testade HipMCL omfattande på Intel Haswell, Ivy Bridge och Knights Landing-processorer på NERSC, använder en upp till 2, 000 noder och en halv miljon trådar på alla processorer, och i alla dessa körningar klustrade HipMCL framgångsrikt nätverk som omfattar tusentals till miljarder kanter, " säger Buluç. "Vi ser att det inte finns någon barriär i antalet processorer som den kan använda för att köra och upptäcker att den kan klustera nätverk 1, 000 gånger snabbare än den ursprungliga MCL-algoritmen."
"HipMCL kommer att bli riktigt transformerande för beräkningsbiologi av big data, precis som IMG- och IMG/M-systemen har varit för mikrobiomgenomik, " säger Kyrpides. "Denna prestation är ett bevis på fördelarna med tvärvetenskapligt samarbete vid Berkeley Lab. Som biologer förstår vi vetenskapen, men det har varit så ovärderligt att kunna samarbeta med datavetare som kan hjälpa oss att ta itu med våra begränsningar och driva oss framåt."
Deras nästa steg är att fortsätta att omarbeta HipMCL och andra beräkningsbiologiska verktyg för framtida exascale-system, som kommer att kunna beräkna kvintiljonberäkningar per sekund. Detta kommer att vara viktigt eftersom genomikdata fortsätter att växa i en häpnadsväckande takt - fördubblas ungefär var femte till var sjätte månad. Detta kommer att göras som en del av DOE Exascale Computing Projects Exagraph co-design center.