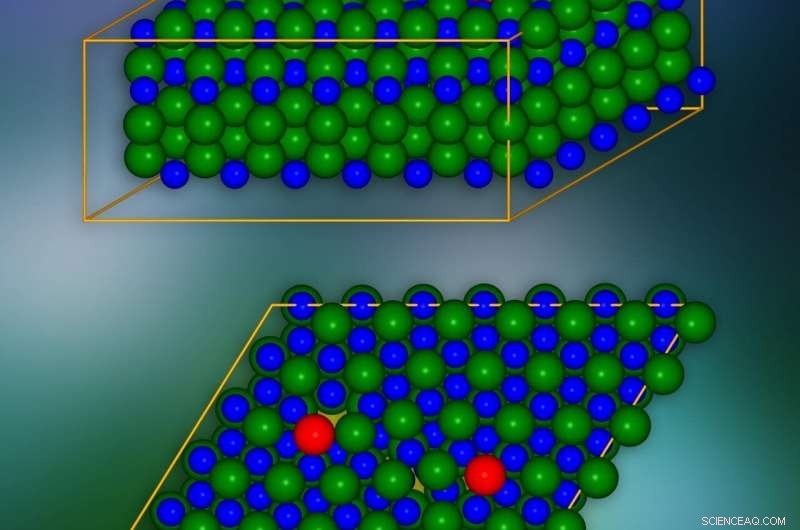
Kiselkarbidkristallmodell med kantförskjutningar införda på platser markerade med rött. Ett enda kristallografiskt plan visas längst ner. De platser där elektriska laddningar kan "läcka" till angränsande lager är markerade med gult. Kredit:IFJ PAN
Brister i kristallstrukturen, särskilt kantförskjutningar av långsträckt natur, djupt modifiera grundläggande egenskaper för hela materialet och, följaktligen, begränsar dess tillämpningar drastiskt. Använd kiselkarbid som exempel, fysiker från Krakow och Warszawa har visat att även sådana beräkningskrävande defekter framgångsrikt kan undersökas med atomnoggrannhet med hjälp av en smart konstruerad, liten i storlek, modell.
Matematik älskar perfektion. Tyvärr, perfektion älskar inte den fysiska verkligheten. Teoretiker som modellerar kristaller har länge försökt inkludera defekter i verkliga kristallina strukturer och förutsäga deras inverkan på materialens fysikaliska egenskaper. Modellerna, baserat på resultaten från olika experiment, har beskrivit förändringar i ett materials grundläggande egenskaper utan att förklara de verkliga orsakerna och effekterna av de förekommande fenomenen.
En ny modell av kiselkarbid (SiC), byggd av fysiker från Institute of Nuclear Physics of the Polish Academy of Sciences (IFJ PAN) i Krakow, har tillåtit dem att visa att det nu är möjligt att studera kristaller ab initio med så komplexa defekter som kantförskjutningar och förklara deras egenskaper genom processer som sker i atomskala. Detta spektakulära resultat, presenterades nyligen vid konferensen Multiscale Phenomena in Molecular Matter 2019 i Krakow, uppnåddes av IFJ PAN -fysikerna i samarbete med Institute of Fundamental Technological Research vid Polska vetenskapsakademien och Institutet för högtrycksfysik vid polska vetenskapsakademien, båda ligger i Warszawa.
"Vi försökte hitta de mekanismer som är ansvariga på atomnivå för att sänka nedbrytningsspänningen i kiselkarbidkristaller. Våra initieringsberäkningar leder till en kvalitativ förståelse av problemet och bidrar till att förklara detaljerna i detta fenomen, "säger doktor Jan Lazewski, professor vid IFJ PAN.
Ab initio -beräkningar har nu en lång historia relaterad till Nobelpriset för Walter Kohn och John Pople 1998 (dock har de nyligen införts för simuleringar av linjära kristalldefekter). Denna term används för att beskriva beräkningar som utförs med hjälp av kvantmekaniska ekvationer, stöds endast av kunskap om atomens struktur och kristallernas symmetri. Det finns ingen direkt information från experiment i sådana modeller, vilket innebär att de också kan användas för att analysera material som aldrig har studerats eller ens syntetiserats tidigare. På grund av relativt stor komplikation av problemet, hittills fungerade ab initio -beräkningar, som mest, vid punktfel, relaterade till lediga platser (saknade atomer eller hål i kristallstrukturen) samt tillsatser införda i kristallen.
Det var inte utan anledning som Krakowforskarna använde kiselkarbid. Egenskaperna hos denna halvledare är så intressanta att det tidigare till och med ansågs vara en efterföljare till kisel. Dess bandgap (barriären som laddningen måste övervinna för att komma från valensbandet till ledningsbandet och leda ström) är nästan tre gånger större än i kisel, den tillåtna ledningsdensiteten - dubbelt så stor, förmågan att sprida värme - mer än tre gånger större, och gränsfrekvensen för kristalldrift så många som sex gånger högre. Dessutom, kiselkarbidsystem kan fungera vid temperaturer upp till 650 grader Celsius, medan kiselsystem redan börjar få problem vid 120 grader Celsius. SiC har också en hög smältpunkt, det är svårt, resistent mot syra och strålning. Dess nackdelar inkluderar framför allt priset:medan två-tums kiselskivor kostar bara några dollar, värdet på liknande kiselkarbidskivor når tusentals. Lågkvalitets kiselkarbidkristaller är ett populärt slipmaterial, används även i skottsäkra västar och i bromsskivorna på världens dyraste bilar, som Lamborghini eller Bugatti. Kristaller av hög kvalitet används för att producera speglar för teleskop och i högspänningsanordningar med hög temperaturbeständighet.
På atomnivå, kiselkarbidkristaller består av många platta skikt anordnade ovanpå varandra. Varje lager liknar en honungskaka:det består av sexkantiga celler där kiselkarbidmolekylerna är placerade vertikalt i hörnen. Varje två intilliggande lager kan kombineras på tre sätt. Flerlagers "smörgåsar" med olika layouter skapar så kallade polytyper, varav det finns mer än 250 när det gäller kiselkarbid. Gruppen från IFJ PAN använde 4H-SiC-polymorfen.
"När man modellerar sådana strukturer, ett av huvudproblemen är beräkningskomplexitet. En modell av ren kristall, saknar tillsatser eller dislokationer, kännetecknas av hög symmetri och kan beräknas även på några minuter. För att utföra en beräkning för ett material med förskjutning, vi behöver månader på att arbeta med en dator med hög effekt, "framhåller Dr Pawel Jochym, professor vid IFJ PAN.
Problemen med kantförskjutningar beror på omfattningen av deras inflytande på materialets kristallstruktur. Som en illustration, de kan jämföras med problemet med att dölja ett gap i en rad brickor på ett golv. Mellanrummet kan "kamoufleras" genom att flytta brickorna på intilliggande rader, men defekten förblir alltid synlig. Kantförskjutningar till följd av bristen på hela längder eller områden av atomer/molekyler i enskilda kristallskikt fungerar på samma sätt, påverkar positionerna för atomer och molekyler i många intilliggande lager. Och eftersom dislokationerna kan sträcka sig över långa sträckor, i praktiken inkluderar störningarna som orsakas av dem hela kristallen.
De mest intressanta fenomenen äger rum i dislokationskärnan, dvs i närheten av kanten av det skadade skiktet i kristallnätverket. För att eliminera långsiktiga effekter orsakade av en enda dislokation, och därmed avsevärt minska antalet atomer som övervägs, ett trick användes:en andra dislokation av den motsatta effekten infördes. På det här sättet, effekten av den första dislokationen över längre sträckor kompenserades.
SiC -kristallmodellen bestod av cirka 400 atomer. Simuleringarna visade att i kristallskikten, längs kanten av defektens kärna, "tunnlar" visas i form av kanaler med reducerad laddningstäthet. De sänker potentialbarriären lokalt och får elektriska laddningar att "läcka" från valensbandet. Dessutom, i den förbjudna klyftan, som i isolatorn garanterar brist på elektrisk konduktivitet, förhållanden uppträder som minskar dess bredd och effektivitet för att begränsa laddningsflödet. Det visades att dessa tillstånd härrör från atomer som ligger i dislokationskärnan.
"Situationen kan jämföras med en djup, brant ravin som en ekorre försöker korsa. Om botten av ravinen är tom, ekorren kommer inte till andra sidan. Dock, om det finns ett antal träd i botten som är tillräckligt höga, ekorren kan hoppa över sina toppar till andra sidan av ravinen. I kristallen vi modellerade, ekorrarna är de elektriska laddningarna, valensbandet är en kant av ravinen, ledningsbandet är det andra, och träden är ovannämnda tillstånd associerade med atomerna i dislokationskärnan, "säger professor Lazewski.
Nu när mekanismerna som ansvarar för att sänka tröskeln för energibarriären har blivit kända på atomnivå, det finns ett stort utrymme för experiment. Den föreslagna mekanismen måste verifieras för att kunna använda den för att begränsa det negativa inflytandet av de testade defekterna. Lyckligtvis, det finns redan tekniska möjligheter för detta.
"Framtiden kommer att verifiera om våra idéer kommer att bekräftas i sin helhet. Men vi är övertygade om ödet för vår modell och det presenterade tillvägagångssättet för att simulera kantförskjutningar. Vi vet redan att ab initio -modellen har visat sitt värde i konfrontation med vissa experimentella data, "avslutar prof. Jochym.