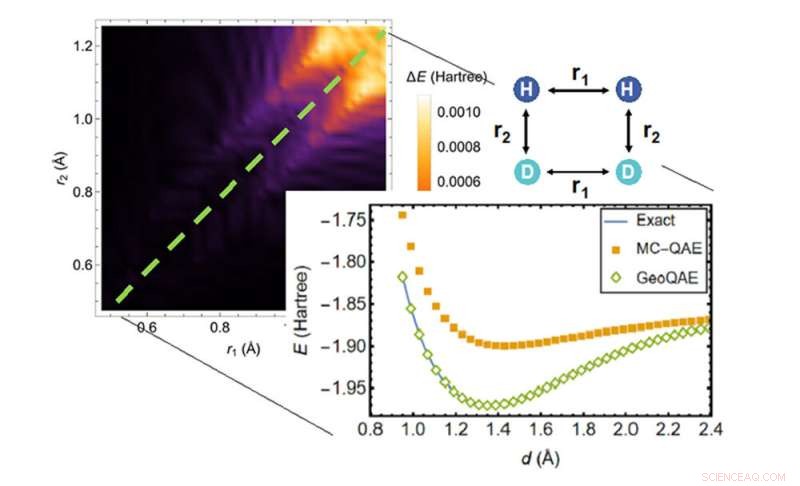
Vid beräkning av den potentiella energiytan för den kemiska reaktionen av H2;+ D2 → 2HD överträffar den nya algoritmen (gröna diamanter) den tidigare algoritmen (orange rutor) när det gäller att hitta den mest exakta lösningen (blå linje). Kredit:Brookhaven National Laboratory
Ett team av forskare från US Department of Energy's (DOE) Brookhaven National Laboratory och Stony Brook University har tagit fram en ny kvantalgoritm för att beräkna de lägsta energierna hos molekyler vid specifika konfigurationer under kemiska reaktioner, inklusive när deras kemiska bindningar bryts. Som beskrivs i Fysisk granskning , jämfört med liknande befintliga algoritmer, inklusive teamets tidigare metod, kommer den nya algoritmen att avsevärt förbättra forskarnas förmåga att exakt och tillförlitligt beräkna den potentiella energiytan i reagerande molekyler.
För detta arbete arbetade Deyu Lu, en Center for Functional Nanomaterials (CFN) fysiker vid Brookhaven Lab, med Tzu-Chieh Wei, en docent specialiserad på kvantinformationsvetenskap vid C.N. Yang Institute for Theoretical Physics vid Stony Brook University, Qin Wu, en teoretiker vid CFN, och Hongye Yu, en Ph.D. student vid Stony Brook.
"Att förstå en molekyls kvantmekanik, hur den beter sig på atomnivå, kan ge nyckelinsikter om dess kemiska egenskaper, som dess stabilitet och reaktivitet", sa Lu.
En speciell egenskap som har varit en utmaning att fastställa är en molekyls grundtillstånd:punkten där molekylens totala elektroniska energi (inklusive kinetisk och potentiell energi) är som lägst och ingenting utanför det "molekylära systemet" är spännande eller laddar molekylens elektroner. När atomstrukturen i ett kemiskt system blir mer komplex, som i en stor molekyl, kan många fler elektroner interagera. Dessa interaktioner gör det extremt svårt att beräkna grundtillståndet för komplexa molekyler.
Den nya kvantalgoritmen förbättrar den tidigare algoritmen för att tackla detta problem på ett kreativt sätt. Den utnyttjar en jämn, geometrisk deformation som görs genom att kontinuerligt variera bindningslängder eller bindningsvinklar i molekylens struktur. Med detta tillvägagångssätt säger forskarna att de kan beräkna grundtillståndet för molekyler mycket exakt, även när kemiska bindningar bryts och reformeras under kemiska reaktioner.
Bygg grunden
"När man enbart förlitar sig på traditionella beräkningsmetoder, innehåller detta grundtillståndsproblem för många variabler att lösa – även på de mest kraftfulla superdatorerna", sa Lu.
Du kan tänka på en algoritm som en uppsättning steg för att lösa ett visst problem. Klassiska datorer kan köra komplexa algoritmer, men när de blir större och mer involverade kan de bli för svåra eller tidskrävande för klassiska datorer att lösa. Kvantdatorer kan påskynda processen genom att utnyttja kvantmekanikens regler.
I klassisk beräkning lagras data i bitar som har värdet 1 eller 0. En kvantbit, känd som en qubit, kan ha ett värde över bara 0 eller 1, den kan till och med ha värdet 0 och 1, i en så kallad kvantsuperposition. I princip kan dessa mer "flexibla" qubits lagra en större mängd information än klassiska bitar. Om forskare kan hitta sätt att utnyttja den informationsbärande kapaciteten hos qubits, kan datorkraften expandera exponentiellt med varje ytterligare qubit.
Qubits är dock ganska ömtåliga. De kan ofta gå sönder när information extraheras. När en kvantenhet interagerar med den omgivande miljön kan den generera brus eller störningar som förstör kvanttillståndet. Temperaturförändringar, vibrationer, elektromagnetiska störningar och till och med materialdefekter kan också göra att qubits förlorar information.
För att kompensera för dessa fallgropar utvecklade forskare en hybridlösning som drar fördel av både klassiska datoralgoritmer, som är mer stabila och praktiska.
Lu och Wei började forska om klassiska hybrid- och kvantberäkningsmetoder 2019. Detta årliga anslag främjar samarbete mellan Brookhaven National Laboratory och Stony Brook University genom att finansiera gemensamma forskningsinitiativ som ligger i linje med båda institutionernas uppdrag. Med detta inledande arbete fokuserade Lu och Wei först på att lösa grundtillståndsproblemet genom att ersätta de "dyraste" klassiska algoritmerna – de som var mycket mer komplexa och krävde betydligt fler steg (och mer beräkningstid) att slutföra – med kvantalgoritmer .
Sträcka band, skapa nya vägar
Forskarna noterar att befintliga kvantalgoritmer alla har nackdelar för att lösa grundtillståndsproblemet, inklusive den som Wei och Yu utvecklade 2019. Medan vissa populära algoritmer är korrekta när en molekyl är i sin jämviktsgeometri - dess naturliga arrangemang av atomer i tre dimensioner - dessa algoritmer kan bli opålitliga när de kemiska bindningarna bryts på stora atomavstånd. Bindningsbildning och dissociation spelar en roll i många tillämpningar, som att förutsäga hur mycket energi som krävs för att få igång en kemisk reaktion, så forskare behövde ett sätt att ta itu med detta problem när molekyler reagerar. De behövde nya kvantalgoritmer som kan beskriva bindningsbrott.
För den här nya versionen av algoritmen arbetade teamet med det Brookhaven-Lab-ledda Co-design Center for Quantum Advantage (C2QA), som bildades 2020. Wei bidrar till centrets mjukvarukraft, som är specialiserad på kvantalgoritmer. Teamets nya algoritm använder en adiabatisk metod – en som gör gradvisa förändringar – men med vissa anpassningar som säkerställer att den förblir tillförlitlig när kemiska bindningar bryts.
"En adiabatisk process fungerar genom att gradvis anpassa villkoren för ett kvantmekaniskt system," förklarade Lu. "På sätt och vis når du en lösning i mycket små steg. Du utvecklar systemet från en enkel, lösbar modell till det slutliga målet, vanligtvis en svårare modell. Utöver grundtillståndet, dock ett många elektroniskt system har många exciterade tillstånd vid högre energier. Dessa exciterade tillstånd kan utgöra en utmaning när man använder den här metoden för att beräkna grundtillståndet."
Wei jämförde en adiabatisk algoritm med att köra längs en motorväg, "om du reser från en stad till nästa finns det flera vägar dit, men du vill hitta den säkraste och mest effektiva."
När det gäller kvantkemi är nyckeln att hitta ett tillräckligt stort "energigap" mellan grundtillståndet och exciterade tillstånd där inga elektrontillstånd existerar. Med ett tillräckligt stort mellanrum kommer fordonen i motorvägsmetaforen inte att "korsa körfält", så deras vägar kan spåras exakt.
"En stor lucka betyder att du kan åka snabbare, så på sätt och vis försöker du hitta en mindre trång motorväg för att köra snabbare utan att träffa någonting", sa Wei.
"Med dessa algoritmer är ingången till vägen en väldefinierad, enkel lösning från klassisk datoranvändning," noterade Wei. "Vi vet också var utgången är - molekylens grundtillstånd - och vi försökte hitta ett sätt att ansluta den till ingången på det mest naturliga sättet, en rak linje.
"Vi gjorde det i vår första tidning, men den raka linjen hade vägspärrar orsakade av att energigapet stängdes och vägar korsades. Nu har vi en bättre lösning."
När forskarna testade algoritmen visade de att även med ändliga bindningslängdsändringar fungerade den förbättrade versionen fortfarande exakt för grundtillståndet.
"Vi gick utanför vår komfortzon, eftersom kemi inte är vårt fokus", sa Wei. "Men det var bra att hitta en sådan här ansökan och främja den här typen av samarbete med CFN. Det är viktigt att ha olika perspektiv i forskningen."
Han noterade den ackumulerade ansträngningen från många människor. "I det stora systemet tror jag att vi gör ett litet bidrag, men det här kan vara en grund för annat arbete inom dessa områden," sa han. "Denna forskning är inte bara grundläggande, utan en bra illustration av hur olika institutioner och anläggningar kan mötas för att utnyttja sina expertområden." + Utforska vidare