Ungefär som människorna som skapade dem, har datorer fysik svårt, men kvantmekaniken är ännu svårare. Men en ny teknik skapad av tre forskare från University of Chicago gör det möjligt för datorer att simulera vissa utmanande kvantmekaniska effekter i komplexa elektroniska material med mycket mindre ansträngning.
Genom att göra dessa simuleringar mer exakta och effektiva hoppas forskarna att tekniken kan hjälpa till att upptäcka nya molekyler och material, till exempel nya typer av solceller eller kvantdatorer.
"Detta framsteg har en enorm potential för att främja vår förståelse av molekylära fenomen, med betydande implikationer för kemi, materialvetenskap och relaterade områden", säger vetenskapsmannen Daniel Gibney, doktorand vid University of Chicago. student i kemi och första författare på tidningen, publicerad 14 december i Physical Review Letters .
Ett löv eller en solpanel ser smidig och enkel ut från utsidan, men zooma ner till den molekylära nivån och du kommer att se en väldigt komplicerad dans av elektroner och molekyler.
För att göra nya framsteg inom hållbarhet, tillverkning, jordbruk och många andra områden, modellerar forskare beteendet hos dessa kemiska och molekylära interaktioner. Detta hjälper till att avslöja nya designmöjligheter för framtiden – för allt från nya sätt att binda koldioxid till nya typer av kvantbitar.
Många framsteg har gjorts under de senaste decennierna, men ett av de områden som har varit envist svårt att simulera är när molekylerna börjar uppvisa komplexa kvantmekaniska beteenden som forskare kallar stark korrelation.
Problemet är att när elektroner börjar visa sina mest kvantmekaniska effekter – som att bli "trasslade" – behöver beräkningarna omedelbart mycket mer datorkraft. Även superdatorer kämpar för att hantera konsekvenserna.
En av de vanligaste beräkningarna kallas densitetsfunktionsteori. "Detta är i grunden den mest allestädes närvarande tekniken för att förutsäga elektronisk struktur, men det är i huvudsak en approximation där alla elektroner behandlas som en funktion av en elektron", förklarade David Mazziotti, professor i kemi och senior författare till studien.
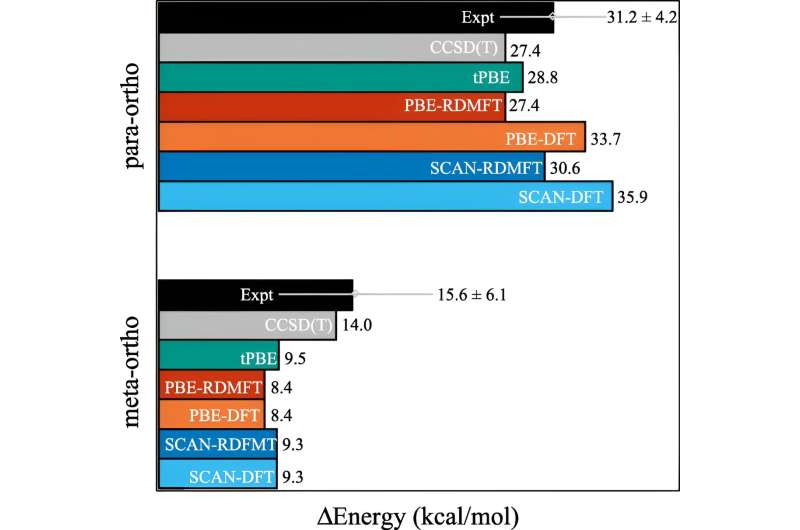
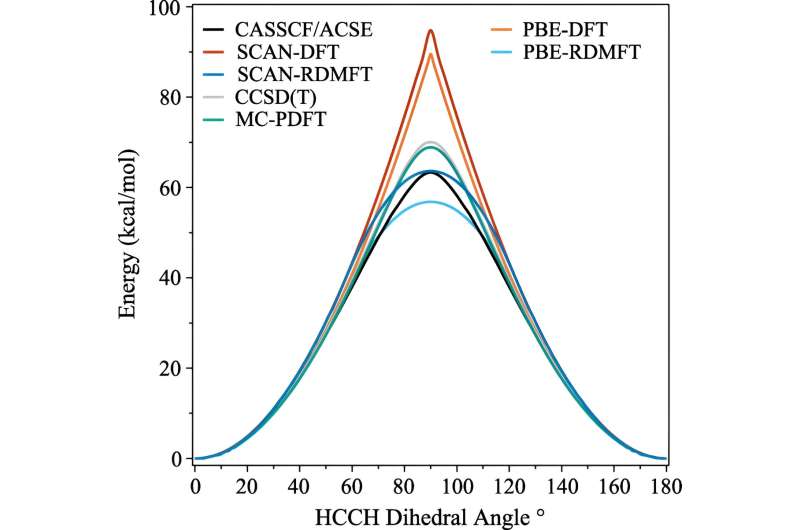
För många beräkningar gör en uppskattning jobbet. Men det börjar gå sönder när elektronernas beteende blir mer korrelerat, vilket händer när kvantmekaniken börjar spela in. Inom kvantmekaniken kan dessa elektroner vara på flera ställen, eller orbitaler, samtidigt. Detta hindrar inte bara mänskliga hjärnor, utan också densitetsfunktionella teorin.
"Och detta är ett viktigt problem, eftersom många av de frågor vi bryr oss om under 2000-talet - som nya molekyler och material för förnybar energi och hållbarhet - kräver att vi utnyttjar materialens kvanta natur", säger Mazziotti.
Mazziotti, Gibney och tredje författare Jan-Niklas Boyn fann att de kunde lägga till en universell korrigering till densitetsfunktionsteorin som gör att elektronerna kan trassla in sig bland flera orbitaler samtidigt.
"Detta gör att orbitalerna i beräkningen inte bara är helt fyllda eller helt tomma, utan var som helst däremellan", sa Mazziotti. "Vi kommer fram till en enelektronbild som fortfarande kan fånga beteendet som uppstår från korrelerade elektroneffekter på många kroppar."
Som en bonus, sa forskarna, kan koden läggas till i befintliga algoritmer utan att behöva skriva om den koden. "I grund och botten sätter korrigeringen igång när den behövs, men stör inte resten av koden annars," sa Gibney.
Det är också universellt – genom att det kan läggas till kod som simulerar många typer av elektroniskt beteende, vare sig det är solcellspaneler eller kolbindning eller supraledande material – eller till och med biologi.
Till exempel, förklarade Boyn, kan en tillämpning vara att förstå kemin som äger rum med hjälp av enzymer som innehåller metallatomer, så kallade metalloenzymer.
"Det finns en uppsjö av metalloenzymer som är ansvariga för mycket av kemin i dina celler, till exempel, men de har varit notoriskt svåra att beskriva med nuvarande modeller," sa han. "Denna teori kan inom en snar framtid tillåta oss att tackla denna kemi på ett sätt som är omöjligt just nu."
Mer information: Daniel Gibney et al, Universal Generalization of Density Functional Theory for Static Correlation, Physical Review Letters (2023). DOI:10.1103/PhysRevLett.131.243003
Journalinformation: Fysiska granskningsbrev
Tillhandahålls av University of Chicago