Stuart Lindsay är chef för Center for Single Molecule Biophysics vid Biodesign Institute vid Arizona Arizona State University. Kredit:Biodesigninstitutet vid Arizona State University
Cirka tre miljarder baspar utgör det mänskliga genomet – livets planlösning. År 2003, Human Genome Project tillkännagav den framgångsrika dekrypteringen av denna kod, en tour de force som fortsätter att tillhandahålla en ström av insikter som är relevanta för människors hälsa och sjukdomar.
Ändå, de primära aktörerna i praktiskt taget alla livsprocesser är de proteiner som kodas för av DNA-sekvenser som kallas gener. För ett brett spektrum av sjukdomar, proteiner kan ge mycket mer övertygande uppenbarelser än vad som kan hämtas från enbart DNA, om forskare kan lyckas låsa upp aminosyrasekvenserna som de är sammansatta av.
Nu, Stuart Lindsay och hans kollegor vid Arizona State Universitys Biodesign Institute har tagit ett stort steg i denna riktning, demonstrerar korrekt identifiering av aminosyror, genom att kort fästa var och en i en smal korsning mellan ett par flankerande elektroder och mäta en karakteristisk kedja av strömspikar som passerar genom på varandra följande aminosyramolekyler.
Genom att använda en maskininlärningsalgoritm, Lindsay och hans team kunde träna en dator att känna igen utbrott av elektrisk aktivitet som representerar den momentana bindningen av en aminosyra i korsningen. Brussignalerna visade sig fungera som pålitliga fingeravtryck, identifiera aminosyror, inklusive subtilt modifierade varianter.
Proteiner tillhandahåller redan en mängd information som är relevant för sjukdomar inklusive cancer, diabetes och neurologiska sjukdomar som Alzheimers, samt ge viktiga insikter i en annan proteinmedierad process:åldrande.
Det nya arbetet främjar möjligheterna till klinisk proteinsekvensering och upptäckten av nya biomarkörer – tidig varning som signalerar sjukdom. Ytterligare, proteinsekvensering kan radikalt förändra patientbehandlingen, möjliggör noggrann övervakning av sjukdomssvar på terapeutika, på molekylär nivå.
Gruppens forskningsresultat redovisas i den avancerade nätupplagan av tidskriften Naturens nanoteknik .
Från genom till proteom
Ett enormt bibliotek av proteiner – känt som proteomet, står i centrum i praktiskt taget alla livsprocesser. Proteiner är avgörande för celltillväxt, differentiering och reparation; de katalyserar kemiska reaktioner och ger försvar mot sjukdomar, bland otaliga hushållsfunktioner.
En av de märkligaste överraskningarna som framkommit från Human Genome Project är det faktum att endast cirka 1,5 procent av genomet kodar för proteiner. Resten av DNA-nukleotiderna bildar regulatoriska sekvenser, icke-kodande RNA-gener, introner, och icke-kodande DNA, (en gång hånfullt märkt "skräp-DNA"). Detta lämnar människor med knappa 20-25, 000 gener, en nykter upptäckt med tanke på att den låga spolmasken har ungefär samma antal. Som professor Lindsay noterar, nyheterna blir värre:"En liljeväxt har ungefär en storleksordning fler gener än vi, " han säger.
Mysteriet med komplexa organismer som människor som bär ett fruktansvärt lågt genantal har att göra med det faktum att proteiner som genereras från DNA-ritningen kan modifieras på ett antal sätt. Faktiskt, forskare har redan identifierat över 100, 000 mänskliga proteiner och forskare som Lindsay tror att detta bara kan vara toppen av isberget.
Precis som meningar kan få sina betydelser ändrade genom ändringar i ordföljd eller skiljetecken, proteiner som genereras från genmallar kan ändra funktion (eller ibland göras obrukbara), ofta med allvarliga konsekvenser för människors hälsa. Två nyckelprocesser som modifierar proteiner är kända som alternativ splitsning och posttranslationell modifiering. De är drivkrafterna bakom den observerade extraordinära proteinvariationen.
Alternativ splitsning sker när kodande regioner av RNA, (kända som exoner) skarvas ihop och icke-kodande regioner (kända som introner) klipps ut, före översättning till proteiner. Denna process sker inte alltid prydligt, med tillfälliga överlappningar av exoner eller introner som introduceras, producerar alternativt splitsade proteiner, vars funktion kan ändras.
Post-translationella modifieringar är markörer som läggs till efter att proteiner har tillverkats. Det finns många former av post-translationell modifiering, inklusive metylering och fosforylering. Vissa förändrade proteiner utför vitala funktioner, medan andra kan vara avvikande och associerade med sjukdom (eller sjukdomsbenägenhet). Ett antal cancerformer är förknippade med sådana proteinfel, som redan används som diagnostiska markörer. Korrekt identifiering av sådana proteiner är dock fortfarande en stor utmaning inom biomedicin.
Nya sekvenser
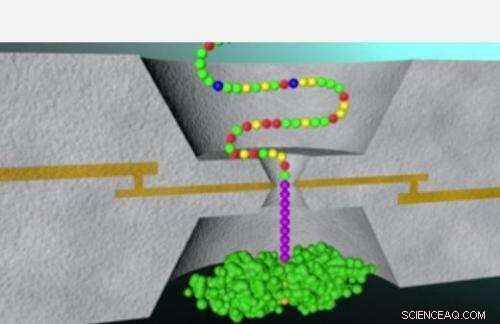
Tekniken som beskrivs i den aktuella forskningen har tidigare tillämpats i Lindsay-labbet för framgångsrik sekvensering av DNA-baser. Denna metod – känd som igenkänningstunnelering – innebär att man trär en peptid genom en liten ögla som kallas en nanopor. Ett par metallelektroder, åtskilda av ett mellanrum på ungefär två nanometer, sitter på vardera sidan av nanoporen när successiva enheter av en peptid träs genom den lilla öppningen, med varje enhet som slutför en elektrisk krets och avger en skur av strömspikar.
Forskargruppen visade att nära analyser av dessa nuvarande toppar kunde göra det möjligt för forskare att avgöra vilken av de fyra nukleotidbaserna - adenin, tymin, cytosin eller guanin - placerades mellan elektroderna i nanoporen.
"Under ett av våra labbmöten för ungefär två år sedan, det föreslogs att kanske samma teknik skulle fungera för aminosyror, ", säger Lindsay. Så började ansträngningarna att ta itu med den betydligt större utmaningen att använda igenkänningstunnel för att identifiera alla 20 aminosyror som finns i proteiner, i motsats till bara 4 baser som består av DNA.
Enkelmolekylsekvensering av proteiner är av enormt värde, offering the potential to detect diminishingly small quantities of proteins that may have been tweaked by alternative splicing or post-translational modification. Ofta, these are the very proteins of interest from the standpoint of recognizing disease states, though current technologies are inadequate to detect them.
As Lindsay notes, there is no equivalent in the protein world to polymerase chain reaction (PCR) technology, which allows minute quantities of DNA in a sample to be rapidly amplified. "We probably don't even know about most of the proteins that would be important in diagnostics. It's just a black hole to us because the concentrations are too low for current analytical techniques, " he says, adding that the ability of recognition tunneling to pinpoint abnormalities on a single molecule basis "could be a complete game changer in proteomics."
The new paper describes a series of experiments in which pure samples of individual amino acids, individual molecules in mixed solution and finally, short peptide chains were successfully identified through recognition tunneling. The work sets the stage for a method to sequence individual protein molecules rapidly and cheaply (see accompanying animation).
A machine learning algorithm known as Support Vector Machine was used to train a computer to analyze the burst signals produced when amino acids formed bonds in the tunnel junction and emitted a lively noise signal as the poised electrodes passed tunneling current through each molecule. (The machine learning algorithm is the same one used by the IBM computer 'Watson' to defeat a human opponent in Jeopardy.)
Lindsay says that around 50 distinct signal burst characteristics were used in the amino acid identifications, but that most of the discriminatory power is achieved with 10 or fewer signal traits.
Anmärkningsvärt, recognition tunneling not only pinpointed amino acids with high reliability from single complex burst signals, but managed to distinguish a post-translationally modified protein (sarcosine) from its unmodified precursor (glycine) and also to discriminate between mirror-image molecules knows as enantiomers and so-called isobaric molecules, which differ in peptide sequence but exhibit identical masses.
Pathway to the $1000 dollar proteome?
Lindsay indicates that the new studies, which rely on innovative strategies for handling single molecules coupled with startling advances in computing power, open up horizons that were inconceivable only a short time ago. It is becoming clear that the tools that made the $1000 genome feasible are equally applicable to an eventual $1000 dollar proteome. Verkligen, such a landmark may not be far off. "Varför inte?" Lindsay asks. "People think it's crazy but the technical tools are there and what will work for DNA sequencing will work for protein sequencing."
While the tunneling measurements have until now been made using a complex laboratory instrument known as a scanning tunneling microscope (STM), Lindsay and his colleagues are currently working on a solid state device capable of fast, cost-effective and clinically applicable recognition tunneling of amino acids and other analytes. Eventual application of such solid-state devices in massively parallel systems should make clinical proteomics a practical reality.