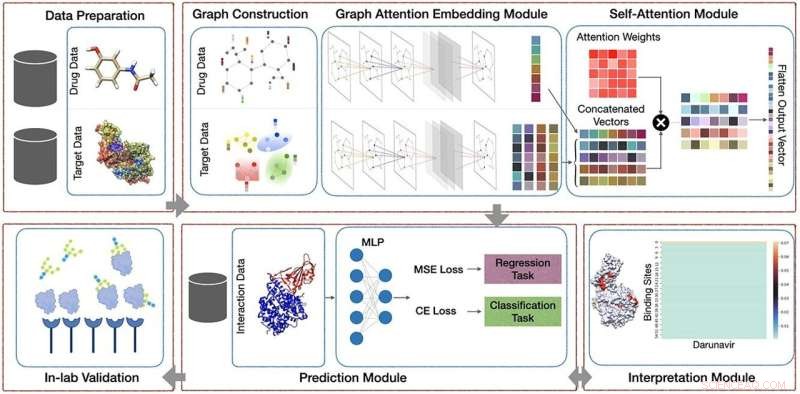
Vårt föreslagna ramverk inkluderar fem huvudmoduler:(1) Förbearbetningsmodul som består av att hitta proteiners bindningsställen; (2) AttentionSiteDTI djupinlärningsmodul, där vi konstruerar grafrepresentationer av liganders SMILE och proteiners bindningsställen, och vi skapar ett grafkonvolutionellt neuralt nätverk beväpnat med en uppmärksamhetspoolmekanism för att extrahera inlärbara inbäddningar från grafer, såväl som en själv- uppmärksamhetsmekanism för att lära sig förhållandet mellan ligander och proteiners bindningsställen; (3) Prediktionsmodul för att förutsäga okänd interaktion i ett läkemedel-målpar, som kan hantera både klassificerings- och regressionsuppgifter; (4) Tolkningsmodul för att ge en djupare förståelse för vilka bindningsställen för ett målprotein som är mer sannolikt att binda med en given ligand. (5) Valideringar i laboratoriet, där vi jämför våra beräkningsmässigt förutsagda resultat med experimentellt observerade (uppmätta) läkemedel-mål-interaktioner i laboratoriet för att testa och validera den praktiska potentialen hos vår föreslagna modell. Kredit:Brefings in Bioinformatics (2022). DOI:10.1093/bib/bbac272
Att utveckla livräddande läkemedel kan ta miljarder dollar och årtionden av tid, men University of Central Florida forskare siktar på att påskynda denna process med en ny artificiell intelligens-baserad drogscreeningsprocess som de har utvecklat.
Med hjälp av en metod som modellerar läkemedels- och målproteininteraktioner med hjälp av naturliga språkbehandlingstekniker, uppnådde forskarna upp till 97 % noggrannhet i att identifiera lovande läkemedelskandidater. Resultaten publicerades nyligen i tidskriften Briefings in Bioinformatics .
Tekniken representerar läkemedel-protein-interaktioner genom ord för varje proteinbindningsställe och använder djupinlärning för att extrahera de egenskaper som styr de komplexa interaktionerna mellan de två.
"I och med att AI blir mer tillgängligt har detta blivit något som AI kan ta itu med", säger studiens medförfattare Ozlem Garibay, biträdande professor vid UCF:s avdelning för industriell teknik och ledningssystem. "Du kan prova så många varianter av proteiner och läkemedelsinteraktioner och ta reda på vilka som är mer benägna att binda eller inte."
Modellen de har utvecklat, känd som AttentionSiteDTI, är den första som kan tolkas med hjälp av språket för proteinbindningsställen.
Arbetet är viktigt eftersom det kommer att hjälpa läkemedelsdesigners att identifiera kritiska proteinbindningsställen tillsammans med deras funktionella egenskaper, vilket är nyckeln till att avgöra om ett läkemedel kommer att vara effektivt.
Forskarna uppnådde framgången genom att ta fram en självuppmärksamhetsmekanism som får modellen att lära sig vilka delar av proteinet som interagerar med läkemedelsföreningarna, samtidigt som den uppnår toppmoderna förutsägelseprestanda.
Mekanismens självuppmärksamhetsförmåga fungerar genom att selektivt fokusera på de mest relevanta delarna av proteinet.
Forskarna validerade sin modell med hjälp av laboratorieexperiment som mätte bindningsinteraktioner mellan föreningar och proteiner och jämförde sedan resultaten med de som deras modell förutspådde beräkningsmässigt. Eftersom läkemedel för att behandla covid fortfarande är intressanta inkluderade experimenten även testning och validering av läkemedelsföreningar som skulle binda till ett spikprotein från SARS-CoV2-viruset.
Garibay säger att den höga överensstämmelsen mellan labbresultaten och beräkningsförutsägelserna illustrerar potentialen hos AttentionSiteDTI att förhandsgranska potentiellt effektiva läkemedelsföreningar och påskynda utforskningen av nya läkemedel och återanvändningen av befintliga.
"Denna forskning med hög effekt var endast möjlig på grund av tvärvetenskapligt samarbete mellan materialteknik och AI/ML och datavetare för att ta itu med COVID-relaterade upptäckter", säger Sudipta Seal, studiemedförfattare och ordförande för UCF:s institution för materialvetenskap och teknik.
Mehdi Yazdani-Jahromi, doktorand vid UCF:s College of Engineering and Computer Science och studiens huvudförfattare, säger att arbetet introducerar en ny riktning i förhandsscreening av läkemedel.
"Detta gör det möjligt för forskare att använda AI för att identifiera läkemedel mer exakt för att reagera snabbt på nya sjukdomar", säger Yazdani-Jahromi. "Denna metod gör det också möjligt för forskarna att identifiera det bästa bindningsstället för ett virusprotein att fokusera på i läkemedelsdesign."
"Nästa steg i vår forskning kommer att vara att designa nya läkemedel med hjälp av kraften i AI", säger han. "Detta kan naturligtvis vara nästa steg för att vara beredd på en pandemi." + Utforska vidare