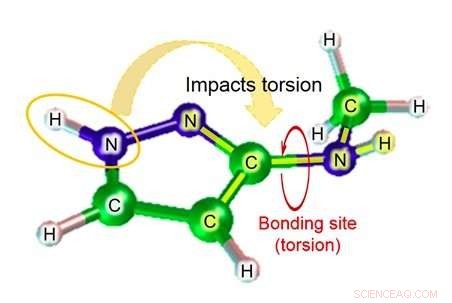
Figur 1:Dihedral vinkel (vinkeln som bildas av planet skapat av atomer A, B, och C, och planet skapat av atomer B, C, och D). Upphovsman:Fujitsu
Fujitsu Laboratories meddelade idag utvecklingen av molekylär simuleringsteknik för läkemedelsupptäckt som exakt kan uppskatta bindningsaffinitet, vilket representerar i vilken grad proteiner som kan orsaka sjukdomar (målproteiner) binder till kemiska ämnen som kan bli kandidatläkemedel. I processen för upptäckt av läkemedel, det finns ett krav på exakt förutsägelse av bindningsaffiniteten mellan målproteiner och kemiska ämnen, som ger en grov uppskattning av ett läkemedels effekt. Molekylär simuleringsteknik har använts i stor utsträckning tidigare som en metod för att förutsäga bindningsaffinitet, beräkna de ungefärliga krafterna som uppstår mellan atomer i molekyler med hjälp av newtonsk mekanik. Problemet med denna metod, dock, kvarstår att den låga gradens noggrannhet av dess uppskattning av de viktigaste parametrarna-graden av vridning vid bindningsställena. Detta innebär att noggrannheten i dess uppskattning av den totala bindningsaffiniteten också är dålig.
Nu, Fujitsu Laboratories har utvecklat molekylär simuleringsteknik som uppskattar vridningsgraden i ett kemiskt ämne, som är direkt kopplad till den förutsagda bindningsaffiniteten. Den nya tekniken tar inte bara hänsyn till bindningsplatsen där torsionen kommer att inträffa, men också effekterna av närliggande atomer. Fujitsu Laboratories utvärderade denna teknik för 190 typer av kemiska ämnen, att jämföra resultaten med korrekta resultat som kommit fram till från första principberäkningen och sedan utvärdera felprocenten. När du gör det, det kunde bekräfta att felprocenten i uppskattningen av vridningsgraden var, i genomsnitt, en tiondel av tidigare teknik. Det förväntas att användningen av denna nya teknik för IT-baserad upptäckt av läkemedel, med sin förmåga att exakt uppskatta bindningsaffiniteten för riktade proteiner och kemiska ämnen, erbjuder potential för banbrytande nya läkemedelsupptäckningsinsatser som inte kunde uppnås med tidigare tillvägagångssätt.
Upptäckten av nya läkemedel kräver betydande utgifter och tidsramar som kan mätas på årtionden, vilket leder till en global sökning efter nya metoder för att upptäcka droger. En av metoderna som har fått stort intresse är IT-baserad upptäckt av läkemedel, en ny metod för upptäckt av läkemedel med hjälp av datorer som gör det möjligt att skapa kemiska ämnen som kandidater för nya läkemedel med stor sannolikhet för framgång. IT-baserad upptäckt av läkemedel har blivit en fokuspunkt för förväntningar som en banbrytande teknik för skapandet av nya läkemedel, för till skillnad från tidigare metoder för försök och fel, där kemiska ämnen upprepade gånger skapas och testas, detta tillvägagångssätt gör det möjligt att i princip utforma kemiska ämnen och uppskatta deras effekter.
Figur 2:Exempel på molekylstruktur:3- (metylamino) pyrazol. Upphovsman:Fujitsu
Effekterna av en kemisk substans som ett läkemedel uttrycks när den kemiska substansen binder till ett målprotein. När den kemiska substansen binder till målproteinet, den kan ändra sin form i linje med målproteinets. Graden av deformation, nämligen, parametrarna som anger omfattningen av denna formförändring, är direkt kopplad till ämnets och proteinets bindningsaffinitet, och ger en grov uppfattning om dess effekt som läkemedel. Med tanke på detta, det finns en stark efterfrågan på förmågan att exakt förutsäga detta värde. För att beräkna graden av deformation av ett kemiskt ämne, det finns metoder baserade på kvantmekanik och metoder baserade på newtonsk mekanik. Kvantmekanikbaserad första principberäkning möjliggör extremt noggranna beräkningar, lösa elektronernas tillstånd från typerna och positionerna för de inblandade atomerna. Å andra sidan, dock, förmågan hos de första principerna att utföra krävande beräkningar leder nödvändigtvis till massiv tid som krävs för att slutföra beräkningarna. För att simulera graden av deformation för många kemiska ämnen, den tid som krävs är i storleksordningen år, gör denna metod opraktisk. Å andra sidan, ungefärliga beräkningar baserade på molekylära simuleringar är extremt snabba, med hjälp av newtonsk mekanik för att beräkna krafterna mellan atomerna i molekylerna, och kan till och med hantera stora molekyler som proteiner ganska enkelt. Följaktligen, denna metod används i stor utsträckning. Med Newtonsk mekanik, krafterna mellan atomerna uttrycks på följande sätt:
Bland dessa, när en kemisk substans är bunden till ett målprotein, graden av vridning av bindningen representerar den viktiga graden av deformation. Med befintlig teknik, dock, noggrannheten i uppskattningen av parametern dihedral vinkel (figur 1), som är nödvändig för att beräkna graden av vridning av bindningen, är ganska låg, vilket resulterar i problemet med låg noggrannhet i uppskattningen av bindningens affinitet i simuleringen.
Fujitsu Laboratories har utvecklat molekylär simuleringsteknik i mer än tio år. Nu, använder den kunskap som den erhållit genom tidigare insatser, Fujitsu Laboratories har utvecklat en molekylär simuleringsteknik som kan uppskatta parametern för dihedral vinkel genom att ta hänsyn till effekterna av atomer nära bindningen. Befintlig teknik uppskattar parametern dihedral vinkel baserat på totalt fyra atomer-de två atomerna i den relevanta bindningen, och de andra atomerna var och en av dessa atomer var bundna till. Beroende på molekylens struktur, dock, det finns fall där atomer utöver de fyra kan ha en betydande inverkan, och i dessa fall, uppskattningens felmarginal kan vara ganska stor. Med denna teknik, Fujitsu Laboratories har skapat en databas med uppskattningsformler för partiella strukturmönster där effekterna av atomer längre bort från bindningsstället kan vara betydande, samt för graden av vridning av kemiska ämnen som kan förväntas i så fall. Med hjälp av den relevanta uppskattningsformeln för att hitta graden av torsion (figur 2) för molekyler som motsvarar databasen för partiella strukturer, det har blivit möjligt att till och med göra mycket exakta uppskattningar för molekylär torsion, som tidigare var svårt att beräkna exakt.
När Fujitsu Laboratories integrerade denna teknik i programvaran som den hade utvecklat för att generera sofistikerade parametrar för krafterna mellan atomer (FF-FOM), den kunde bekräfta att resultaten överensstämde med exakta beräkningar.

Figur 3:Utvärdering av prestandan för dihedrala vinkelparametervärden med 190 typer av kemiska sammansatta strukturer. Upphovsman:Fujitsu
När Fujitsu Laboratories utvärderade skillnaden mellan resultaten av denna teknik och resultaten från en beräkning från de första principerna för uppskattning av vridningsgraden med 190 typer av kemiska ämnen, det var mindre än en tiondel av den tidigare tekniken, i genomsnitt, 0,6 kcal/mol under termiska fluktuationer vid rumstemperatur, bekräftar att den nya tekniken är praktisk. Eftersom den exakt kan uppskatta bindningsaffiniteten för målproteiner och kemiska ämnen, det förväntas att användningen av denna teknik kommer att leda till skapandet av banbrytande nya läkemedel genom dess användning vid IT-baserad upptäckt av läkemedel.