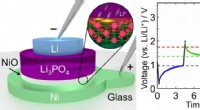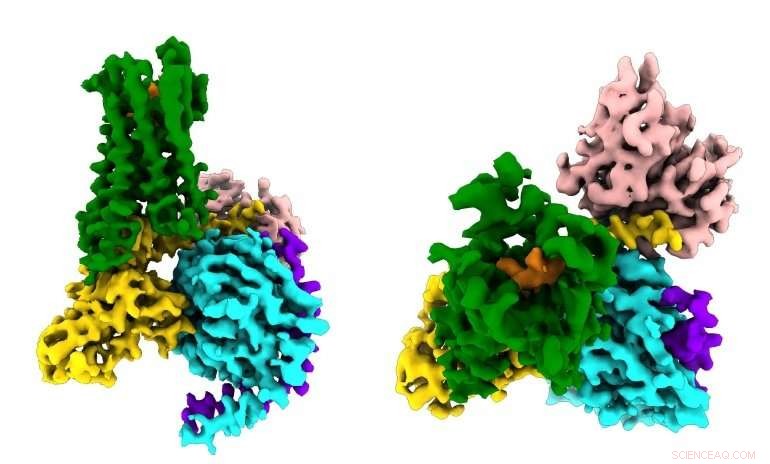
Forskare ser för första gången hur ett syntetiskt opioidläkemedel (apelsin) binder sig till µ-opioidreceptorerna (gröna) i hjärnan, och aktivera signalmolekyler inom neuroner (G⍺ i guld, Gβ i cyan, Gy i lila) som leder till smärtdämpning och missbruk. Upphovsman:Antoine Koehl (Manglik lab)
Opioida läkemedel som morfin och fentanyl är en grundpelare i modern smärtmedicin. Men de orsakar också förstoppning, är mycket beroendeframkallande, och kan leda till dödligt andningssvikt om det tas vid en för hög dos. Forskare har länge försökt utveckla nya opioida läkemedel som kan driva bort smärta utan dessa farliga biverkningar, men hål i vår förståelse av exakt hur opioider utövar sina olika effekter på biologisk nivå har hittills hållit denna dröm på avstånd.
Opioida smärtstillande medel fungerar genom att binda till ett receptorprotein som finns på nervceller som kallas µ-opioidreceptorn, som utvecklats för att reagera på kroppens naturliga smärtstillande medel (t.ex. endorfiner som produceras av träning) genom att stämma smärta och skapa en känsla av eufori. Opioida läkemedel från opium till morfin till heroin kapar detta signalsystem genom att binda till samma receptormolekyl. Men detaljer om hur aktivering av dessa receptorer utlöser läkemedlets positiva och negativa effekter har förblivit disiga.
Nu, i en studie publicerad den 13 juni, 2018 i Natur , forskare vid UC San Francisco och Stanford University har använt ultrahögupplöst kryo-elektronmikroskopi (cryoEM) för att fånga det mest detaljerade porträttet någonsin av ett opioidläkemedel som utlöser den biokemiska signalkaskaden som ger den dess kraft-både på gott och ont .
"Vi har i huvudsak fångat upp denna signalhändelse i akten, "säger studiens medförfattare Aashish Manglik, MD, Ph.D., en biträdande professor i farmaceutisk kemi vid UCSF's School of Pharmacy som genomförde den nya studien som doktorand och Distinguished Fellow vid Stanford. "Dessa nya bilder på atomnivå kommer förhoppningsvis att göra det möjligt för oss att rationellt utforma föreningar som är inriktade på olika aspekter av opioidsignalering i hjärnan, med hopp om att identifiera nya, säkrare smärtstillande. "
Μ-opioidreceptorn är en del av en stor familj med hundratals signalproteiner som kallas G-proteinkopplade receptorer (GPCR) som är involverade i allt från syn och hörsel till immunsystemets svar på invasiva patogener, och är mål för mer än 30 procent av moderna droger. De flesta GPCR har samma grundläggande mekanismer:När rätt signalmolekyl (t.ex. en opioid) binder till en GPCR på utsidan av cellen, proteinet stimulerar en kedjereaktion av biokemiska signaler i cellen genom att aktivera en budbärarmolekyl som kallas ett G -protein (därav namnet GPCR).
Experiment som avslöjade hur en annan typ av GPCR binder till det "stimulerande" G -proteinet ledde till ett Nobelpris för Stanfords Brian Kobilka, MD, en av seniorförfattarna till den nya studien, men forskare har vetat i decennier att GPCR också kan binda till så många som ett dussin andra signalmolekyler i cellen. Till exempel, µ-opioidreceptorer aktiverar vanligtvis bara så kallade "hämmande" G-proteiner, som har motsatt effekt av den stimulerande G -proteinkaskaden. Dock, forskare är inte säkra på vad som orsakar vissa GPCR:s affinitet för specifika partnerproteiner i cellen, eller exakt vad konsekvenserna blir.
Forskare hoppas att genom att förstå dessa olika vägar för GPCR -signalering, de kanske kan utveckla läkemedel med mycket specifika effekter, som att dämpa smärta utan att orsaka missbruk. Men tills nu, forskare hade liten aning om hur en given GPCR selektivt interagerar med endast en delmängd av signalpartners inom cellen.
Den nya studien, publicerad 13 juni, 2018 i Natur , fångade för första gången hur µ-opioidreceptorn binder till sin hämmande G-proteinpartner. Bland andra fynd, studien visade att receptorns selektivitet verkar bero på den lilla storleken på bindfickan för G -proteinet på insidan av cellen, medan det stimulerande G -proteinet kräver ett större bindningsställe.
Manglik samarbetade tidigare med Brian Shoichets beräkningsdrog för läkemedelsupptäckt, Ph.D., professor i farmaceutisk kemi vid UCSF:s farmaceutiska högskola, att identifiera en molekyl som kallas PZM21 som gör att µ-opioidreceptorn endast kan koppla in det hämmande G-proteinet men inte en annan signalmolekyl som kallas beta-arrestin, och visade att detta selektiva läkemedel gav smärtlindring med minskade biverkningar hos möss. Hans laboratorium bygger nu på det nya, högupplöst porträtt av opioidreceptorn - G -proteinkomplex för att utveckla nytt, ännu mer selektiva föreningar.