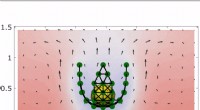
Upphovsman:Caltech
I takt med att kemin har blivit mer avancerad och de kemiska reaktionerna mer komplexa, det är inte längre alltid praktiskt för forskare att sätta sig vid en labbbänk och börja blanda kemikalier för att se vad de kan hitta på.
Tom Miller, en professor i kemi vid Caltech; Matt Welborn, en postdoktor vid Resnick Sustainability Institute; och Lixue Cheng, en doktorand i kemi och kemiteknik, har utvecklat ett nytt verktyg som använder maskininlärning för att förutsäga kemiska reaktioner långt innan reagens träffar provröret.
Deras är inte det första beräkningsverktyget som utvecklats för att göra kemiförutsägelser, men det förbättrar det som redan används, och det spelar roll eftersom den här typen av förutsägelser har stor inverkan på fältet.
"De tillåter oss att ansluta underliggande mikroskopiska egenskaper till de saker vi bryr oss om i den makroskopiska världen, "Miller säger. "Dessa förutsägelser tillåter oss att i förväg veta om en katalysator kommer att prestera bättre än en annan och att identifiera nya läkemedelskandidater."
De kräver också en hel del beräkningsmässigt tunga lyft. Miller påpekar att en betydande del av all superdatortid på jorden är tillägnad kemiförutsägelser, så effektivitetsökningar kan spara forskare mycket tid och kostnader.
Caltech-forskarnas arbete ger i huvudsak en förändring av fokus för förutsägelseprogramvara. Tidigare verktyg baserades på tre beräkningsmodelleringsmetoder kända som densitetsfunktionsteori (DFT), kopplad klusterteori (CC), eller Møller–Plessets störningsteori (MP2). Dessa teorier representerar tre olika tillvägagångssätt för att approximera en lösning på Schrödinger-ekvationen, som beskriver komplexa system där kvantmekaniken spelar en stor roll.
Var och en av dessa teorier har sina egna fördelar och nackdelar. DFT är något av ett snabbt och smutsigt tillvägagångssätt som ger forskarna svar snabbare men med mindre noggrannhet. CC och MP2 är mycket mer exakta men tar längre tid att beräkna och använder mycket mer datorkraft.
Mjölnare, Cheng, och Welborns verktyg trär nålen, ger dem tillgång till förutsägelser som är mer exakta än de som skapats med DFT och på kortare tid än CC och MP2 kan erbjuda. De gör detta genom att fokusera sin maskininlärningsalgoritm på egenskaperna hos molekylära orbitaler – molnet av elektroner runt en molekyl. Redan befintliga verktyg, i kontrast, fokusera på typerna av atomer i en molekyl eller vinklarna vid vilka atomerna är sammanbundna.
Än så länge, deras tillvägagångssätt visar mycket lovande, även om det bara har använts för att göra förutsägelser om relativt enkla system. Det sanna testet, Miller säger, är att se hur det kommer att fungera på mer komplicerade kemiska problem. Fortfarande, han är optimistisk utifrån de preliminära resultaten.
"Om vi kan få det här att fungera, det kommer att vara en stor sak för det sätt på vilket datorer används för att studera kemiska problem, " säger han. "Vi är väldigt exalterade över det."
Arbetet beskrivs i en artikel med titeln "Transferability in Machine Learning for Electronic Structure via the Molecular Orbital Basis" som visas i Journal of Chemical Theory and Computation .