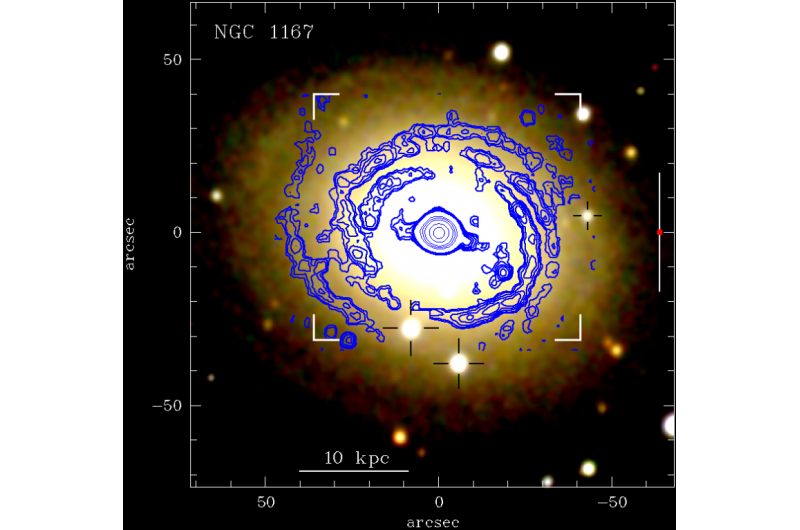
Använder maskininlärning för att hjälpa molekylär design. Kredit:Wenbo Sun, Vetenskapens framsteg, doi:10.1126/sciadv.aay4275
För att syntetisera högpresterande material för organiska solceller (OPV) som omvandlar solstrålning till likström, materialforskare måste på ett meningsfullt sätt fastställa sambandet mellan kemiska strukturer och deras fotovoltaiska egenskaper. I en ny studie om Vetenskapens framsteg , Wenbo Sun och ett team inklusive forskare från School of Energy and Power Engineering, School of Automation, datavetenskap, Elektroteknik och grön och intelligent teknik, etablerat en ny databas med mer än 1, 700 givarmaterial med hjälp av befintliga litteraturrapporter. De använde övervakad inlärning med maskininlärningsmodeller för att bygga struktur-egenskapsrelationer och snabbskärms-OPV-material med hjälp av en mängd olika indata för olika ML-algoritmer.
Använda molekylära fingeravtryck (kodar en struktur av en molekyl i binära bitar) utöver en längd på 1000 bitar Sun et al. erhållit hög ML-prediktionsnoggrannhet. De verifierade tillförlitligheten av metoden genom att screena 10 nydesignade donatormaterial för överensstämmelse mellan modellförutsägelser och experimentella resultat. ML-resultaten presenterade ett kraftfullt verktyg för att förscreena nya OPV-material och påskynda utvecklingen av OPV inom materialteknik.
Organiska fotovoltaiska (OPV) celler kan underlätta direkt och kostnadseffektiv omvandling av solenergi till elektricitet med snabb tillväxt på senare tid för att överstiga energiomvandlingseffektiviteten (PCE). Mainstream OPV-forskning har fokuserat på att bygga en relation mellan nya OPV molekylära strukturer och deras fotovoltaiska egenskaper. Den traditionella processen involverar vanligtvis design och syntes av fotovoltaiska material för montering/optimering av fotovoltaiska celler. Sådana tillvägagångssätt resulterar i tidskrävande forskningscykler som kräver känslig kontroll av kemisk syntes och tillverkning av anordningar, experimentella steg och rening. Den befintliga OPV-utvecklingsprocessen är långsam och ineffektiv med mindre än 2000 OPV-donatormolekyler syntetiserade och testade hittills. Dock, data som samlats in från årtionden av forskningsarbete är ovärderliga, med potentiella värden som återstår att utforska fullt ut för att generera högpresterande OPV-material.
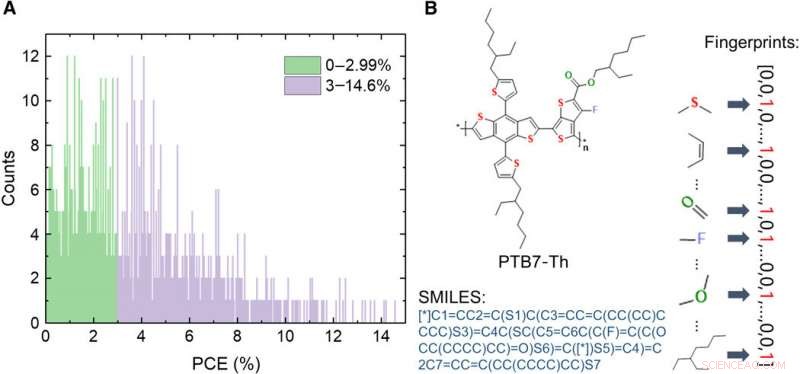
Information om databasen för OPV-givarmaterial. (A) Fördelning av PCE-värden för de 1719 molekylerna i databasen. (B) Schema över uttryck för en molekyl, inklusive bild, förenklat linjeingångssystem med molekylär ingång (SMILES), och fingeravtryck. Kredit:Science Advances, doi:10.1126/sciadv.aay4275
För att extrahera användbar information från data, Sun et al. krävde ett sofistikerat program för att skanna igenom en stor datamängd och extrahera relationer från funktionerna. Eftersom maskininlärning (ML) tillhandahåller beräkningsverktyg för att lära sig och känna igen mönster och samband med hjälp av en träningsdatauppsättning, teamet använde ett datadrivet tillvägagångssätt för att möjliggöra ML och förutsäga olika materialegenskaper. ML-algoritmen behövde inte förstå kemin eller fysiken bakom materialegenskaperna för att utföra uppgifterna. Liknande metoder har nyligen förutspått materials aktivitet/egenskaper framgångsrikt under materialupptäckt, läkemedelsutveckling och materialdesign. Före ML-ansökningar, forskare hade skapat kemiformatik för att skapa en användbar verktygslåda.
Materialforskare har först nyligen utforskat tillämpningarna av ML inom OPV-området. I detta arbete, Sun et al. upprättat en databas som innehåller 1719 experimentellt testade OPV-material från givare som samlats in från litteratur. De studerade vikten av programmeringsspråksuttryck av molekylerna först för att förstå ML-prestanda. De testade sedan flera olika typer av uttryck inklusive bilder, ASCII-strängar, två typer av deskriptorer och sju typer av molekylära fingeravtryck. De observerade att modellförutsägelserna var i god överensstämmelse med de experimentella resultaten. Forskarna förväntar sig att det nya tillvägagångssättet avsevärt påskyndar utvecklingen av nya och högeffektiva organiska halvledande material för OPV-forskningstillämpningar.
Forskargruppen omvandlade först rådata till en maskinläsbar representation. En mängd olika uttryck existerar för samma molekyl som omfattar mycket olika kemisk information presenterad på olika abstrakta nivåer. Med hjälp av en uppsättning ML-modeller, Sun et al. utforskade olika uttryck för en molekyl genom att jämföra deras förutspådda noggrannhet för effektomvandlingseffektivitet (PCE) för att erhålla en djupinlärningsmodellnoggrannhet på 69,41 procent. Den relativt otillfredsställande prestandan berodde på databasens ringa storlek. Till exempel, tidigare när samma grupp använde ett större antal molekyler på upp till 50, 000, noggrannheten för djupinlärningsmodellen översteg 90 procent. För att fullt ut träna en modell för djupinlärning, forskare måste implementera en större databas som innehåller miljontals prover.
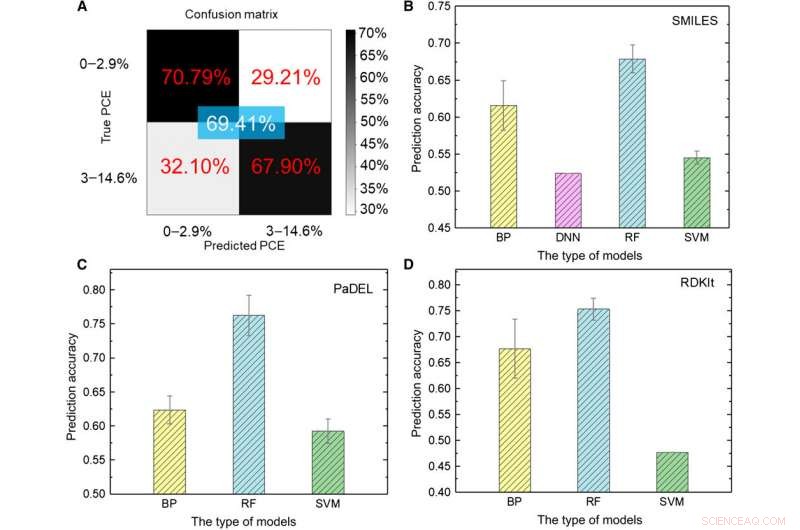
Testresultat av ML-modeller. (A) Testning av djupinlärningsmodellen med bilder som input. (B till D) Testresultat av olika ML-modeller med (B) SMILES, (C) PaDEL, och (D) RDKIt-deskriptorer som indata. Kredit:Science Advances, doi:10.1126/sciadv.aay4275
Sun et al. hade bara hundratals molekyler i varje kategori för närvarande, vilket gör det svårt för modellen att extrahera tillräckligt med information för högre noggrannhet. Även om det är möjligt att finjustera en förtränad modell för att minska mängden data som krävs, tusentals prover är fortfarande nödvändiga för att åstadkomma ett tillräckligt antal funktioner. Detta ledde till möjligheten att öka storleken på databasen när bilder användes för att uttrycka molekyler.
Forskarna använde fem typer av övervakade ML-algoritmer i studien, inklusive (1) backpropagation (BP) neurala nätverk (BPNN), (2) djupt neuralt nätverk (DNN), (3) djupinlärning, (4) stödvektormaskin (SVM) och (5) slumpmässig skog (RF). Dessa var avancerade algoritmer, där BPNN, DNN och djupinlärning baserades på det artificiella neutrala nätverket (ANN). SMILES-koden (förenklat system för inmatning av molekylär inmatning) gav ett annat ursprungligt uttryck för en molekyl, vilket Sun et al. används som ingångar för fyra modeller. Baserat på resultaten, den högsta noggrannheten var ungefär 67,84 procent för RF-modellen. Som förut, till skillnad från djupinlärning, de fyra klassiska metoderna kunde inte extrahera dolda drag. Som helhet, SMILES presterade sämre än bilder som deskriptorer av molekyler för att förutsäga PCE-klassen (power conversion efficiency) i data.
Forskarna använde sedan molekylära deskriptorer som kan beskriva egenskaperna hos en molekyl med hjälp av en rad tal istället för det direkta uttrycket av en kemisk struktur. Forskargruppen använde två typer av deskriptorer PaDEL och RDKIt i studien. Efter omfattande analyser av alla ML-modeller, en stor datastorlek innebar att fler deskriptorer irrelevanta för PCE påverkar ANN-prestandan. Jämförelsevis, en liten datastorlek innebar ineffektiv kemisk information för att effektivt träna ML-modeller, när man använder molekylära deskriptorer som input i ML-metoder, nyckeln förlitade sig på att hitta lämpliga deskriptorer som direkt relaterade till målobjektet.
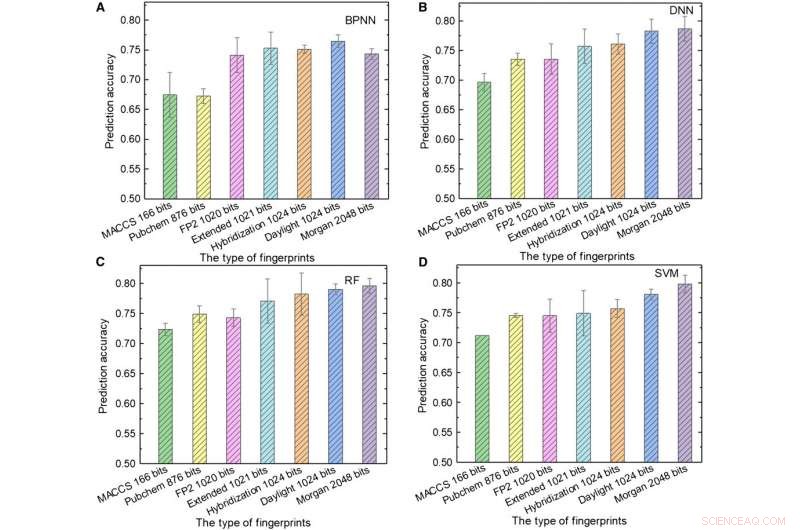
Prestanda för ML-modeller. (A till D) Testresultaten av (A) BPNN, (B) DNN, (C) RF, och (D) SVM använder olika typer av fingeravtryck som indata. Kredit:Science Advances, doi:10.1126/sciadv.aay4275.
Teamet använde sedan molekylära fingeravtryck; typiskt utformade för att representera molekyler som matematiska objekt och ursprungligen skapade för att identifiera isomerer. Under storskalig databasscreening, konceptet representeras som en uppsättning bitar som innehåller "1" s och "0" s för att beskriva närvaron eller frånvaron av specifika understrukturer eller mönster i molekylerna. Sun et al. använde sju typer av fingeravtryck som indata för att träna ML-modellerna och övervägde inverkan av fingeravtryckslängden på prediktionsprestandan hos olika modeller för att få olika fingeravtryck. Till exempel, Molecular Access System (MACCS) fingeravtryck innehöll 166 bitar och var den kortaste ingången och resultaten var otillfredsställande på grund av deras begränsade information.
Sun et al. visade den bästa kombinationen av programmeringsspråk och ML-algoritm erhållen med hybridiseringsfingeravtryck på 1024 bitar och RF, för att uppnå en prediktionsnoggrannhet på 81,76 procent; där Hybridiseringsfingeravtryck representerade SP2-hybridiseringstillstånd för molekyler. När fingeravtryckslängden ökade från 166 till 1024 bitar, prestandan för alla ML-modeller förbättrades eftersom längre fingeravtryck innehöll mer kemisk information.
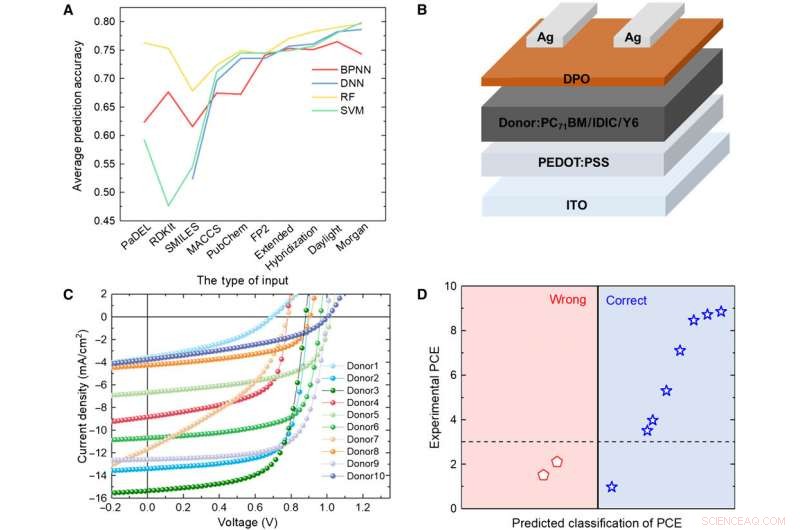
Verifiering av ML-modeller med experiment. (A) Jämförelse av resultaten från fyra olika modeller. (B) Schematiskt diagram över cellarkitekturen som används i denna studie. (C) J-V-kurva för solcellen med det aktiva skiktet med hjälp av det förutsagda givarmaterialet. (D) Förutsägelseresultat kontra experimentella data för de förutspådda givarmaterialen med RF-algoritmen och Daylight-fingeravtryck. Kredit:Science Advances, doi:10.1126/sciadv.aay4275.
För att testa tillförlitligheten hos ML-modellerna, Sun et al. syntetiserade 10 nya OPV-donatormolekyler. Använde sedan tre representativa fingeravtryck för att uttrycka den kemiska strukturen hos de nya molekylerna och jämförde resultaten som förutspåddes av RF-modellen och de experimentella PCE-värdena. Systemet klassificerade åtta av de 10 molekylerna. Resultaten indikerade potentialen hos de syntetiska materialen för OPV-applikationer med ytterligare experimentell optimering för två av de nya materialen. En mindre förändring i strukturen kan orsaka en stor skillnad i PCE-värden. Uppmuntrande, ML-modellerna identifierade sådana mindre modifieringar för att underlätta gynnsamma förutsägelseresultat.
På det här sättet, Wenbo Sun och kollegor använde en litteraturdatabas om OPV-donatormaterial och en mängd olika programmeringsspråksuttryck (bilder, ASCII-strängar, deskriptorer och molekylära fingeravtryck) för att bygga ML-modeller och förutsäga motsvarande OPV PCE-klass. Teamet visade ett schema för att designa OPV-donatormaterial med hjälp av ML-metoder och experimentell analys. De förscreenade ett stort antal donatormaterial med hjälp av ML-modellen för att identifiera ledande kandidater för syntes och ytterligare experiment. Det nya arbetet kan påskynda ny donatormaterialdesign för att påskynda utvecklingen av hög PCE OPV. Användningen av ML i samband med experiment kommer att utveckla materialupptäckten.
© 2019 Science X Network