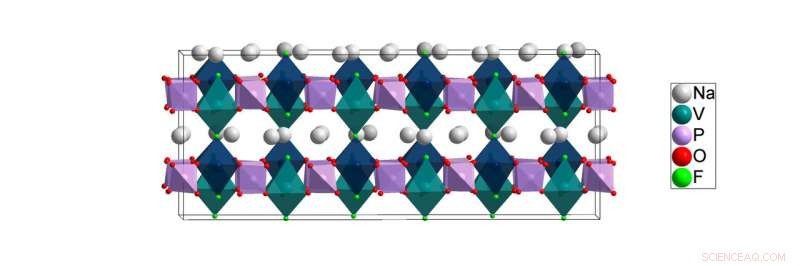
Lågtemperaturstrukturen för NVPF [Na3V2 (PO4) 2F3] löstes i detta arbete. Beräkningar från Lawrence Berkeley National Laboratory tyder på att natriumatomerna (vita) lättast kan röra sig i planen mellan katjonplatserna för vanadin (kricka) och fosfor (mauve) atomer under batterianvändning. Upphovsman:Brookhaven National Laboratory
Forskare vid U.S. Department of Energy's (DOE) Brookhaven National Laboratory, Stony Brook University (SBU), materialprojektet vid DOE:s Lawrence Berkeley National Laboratory (Berkeley Lab), University of California, Berkeley, och europeiska samarbetspartners har utvecklat ett nytt sätt att dechiffrera strukturen på atomnivå för material baserat på data som hämtats från slipade pulverprover. De beskriver deras tillvägagångssätt och visar dess förmåga att lösa strukturen i ett material som visar löften för att skjuta joner genom natriumjonbatterier i ett papper som just publicerats i tidningen Materialkemi .
"Vår metod kombinerar experiment, teori, och moderna beräkningsverktyg för att tillhandahålla strukturella data av hög kvalitet som behövs för att förstå viktiga funktionella material, även om endast pulverprover finns tillgängliga, "sade motsvarande författare Peter Khalifah, som har ett gemensamt möte på Brookhaven Lab och SBU.
Tekniken är på något sätt en form av omvänd teknik. Istället för att lösa strukturen direkt från de experimentella data som mäts på pulverprovet - ett problem som är för komplext för att vara möjligt för många material - använder det datoralgoritmer för att bygga och utvärdera alla materialets troliga strukturer. Genom att analysera "genomet" som är associerat med ett material på detta sätt, det kan vara möjligt att hitta rätt struktur även när denna struktur är så komplex att konventionella metoder för strukturlösning misslyckas.
Frysram för batterikatod
För studien som beskrivs i tidningen, Röntgenpulverdiffraktionsförsök utfördes vid ALBA-synkrotronen i Barcelona, Spanien, av europeiska medarbetare Matteo Bianchini och Francois Fauth, del av ett team som leds av Christian Masquelier. Forskare använde den anläggningens ljusa röntgenstrålar för att studera atomarrangemanget av ett natriumjonbatterikatodmaterial som kallas NVPF vid en mängd olika temperaturer som sträcker sig från rumstemperatur ner till de mycket låga kryogena temperaturerna vid vilka atmosfäriska gaser vätsker. Detta arbete är nödvändigt eftersom störningen i rumstemperaturstrukturen hos NVPF försvinner när det kyls till kryogena temperaturer. Och medan batterierna fungerar nära rumstemperatur, att dechiffrera materialets kryogena struktur är fortfarande kritiskt viktigt eftersom endast denna störningsfria, lågtemperaturstruktur kan ge forskare en klar förståelse av den verkliga kemiska bindningen som finns vid rumstemperatur. Denna kemiska bindningsmiljö påverkar starkt hur joner rör sig genom strukturen vid rumstemperatur och påverkar därmed NVPF:s prestanda som batterimaterial.
"Bindningsmiljön kring natriumatomer - hur många grannar var och en har - är i huvudsak densamma vid låg temperatur som vid rumstemperatur, "Förklarade Khalifah, men att försöka fånga dessa detaljer vid rumstemperatur är som att försöka få barn att sitta stilla för ett foto. "Allt blir suddigt eftersom jonerna rör sig för snabbt för att ta en bild." Av denna anledning, några av de bindningsmiljöer som utgår från rumstemperaturdata är inte korrekta. I kontrast, kryogena temperaturer fryser rörelsen av natriumjoner för att ge en sann bild av den lokala miljön där natriumjonerna sitter när de inte rör sig.
"När materialet kyls, tjugofyra angränsande natriumjoner tvingas var och en att välja en av två möjliga platser, och deras lägsta energiföredragna "beställningsmönster" kan lösas, "Sa Khalifah.
En preliminär analys av pulverröntgendiffraktionsdata av Bianchini indikerade att mönstret för beställning är mycket komplext. För material med så komplexa beställningar, det är vanligtvis inte möjligt att lösa deras tredimensionella atomstruktur med hjälp av pulverdiffraktionsdata.
"Pulverdiffraktionsdata blir platt till en dimension, så mycket information går förlorad, "Sa Khalifah.
Men material gjorda av många olika typer av element, som är fallet för NVPF - som är byggt av natriumatomer, vanadin, fosfor, fluor, och syre med en övergripande kemisk formel för Na 3 V 2 (PO 4 ) 2 F 3 -är för svårt att växa till större kristaller för mer konventionell 3D-röntgenkristallografi.
Så, Brookhaven -gruppen samarbetade med John Dagdelen och andra forskare vid Lawrence Berkeley National Laboratory för att utveckla en ny "genomisk" metod som kan lösa mycket komplexa strukturer med endast pulverdiffraktionsdata. Samarbetsarbetet genomfördes inom Materialprojektet, ett DOE-finansierat forskargrupp under ledning av Kristin Persson vid LBNL som utvecklar innovativa beräkningsmetoder för att påskynda upptäckten av nya funktionella material.
"I stället för att använda pulverdiffraktionsdata för att direkt lösa strukturen, vi tog ett alternativt tillvägagångssätt, "Sa Khalifah." Vi frågade, 'vad är alla troliga arrangemang av natriumjoner i strukturen, "och sedan testade vi var och en av dem automatiskt för att jämföra det med experimentella data för att ta reda på vad strukturen var."
NVPF -strukturen är en av de mest komplexa som någonsin lösts för ett material som endast använder pulverdiffraktionsdata.
"Vi kunde inte ha gjort den här vetenskapen utan moderna beräkningsverktyg - uppräkningsmetoderna som används för att generera de kemiskt trovärdiga strukturerna och de sofistikerade automatiserade skripten för att förfina de strukturer som använde programvarubiblioteket pymatgen (Python Materials Genomics), "Sa Khalifah.
Nollställer strukturen
Baserat på tillgänglig strukturell kunskap för NVPF och på en uppsättning grundläggande kemiska regler för bindning, det finns mer än en halv miljon troliga beställningsmönster för natriumatomerna i NVPF. Även efter att ha använt beräkningsalgoritmer för att identifiera likvärdiga strukturer som genereras genom olika ordningsval, nästan 3, 000 unika möjliga beställningar kvar.
"Dessa 3, 000 försöksstrukturer är mer än vad som rimligen kan testas för hand, men deras riktighet kan utvärderas av en enda dator som arbetar non-stop i ungefär två dagar, "Sa Khalifah.
Korrektheten i varje försöksstruktur utvärderades med hjälp av programvara för att förutsäga hur dess pulverröntgendiffraktionsmönster skulle se ut, och sedan jämföra de beräknade resultaten med de experimentellt uppmätta diffraktionsdata, arbete utfört av Stony Brook Ph.D. studenten Gerard Mattei. Om skillnaden mellan de förutsagda och observerade diffraktionsmönstren är relativt liten, mjukvaran kan optimera alla teststrukturer genom att justera positionerna för dess ingående atomer för att förbättra överensstämmelsen mellan de beräknade och observerade mönstren.
Men även efter en sådan finjustering, nästan 2, 500 av de optimerade strukturerna kan användas för att passa de experimentella diffraktionsdatorna väl.
"Vi förväntade oss inte att få så många bra passningar, "Sa Khalifah." Så, vi hade en andra utmaning att avgöra vilken av de många möjliga strukturerna som var korrekt genom att titta på vilken som hade rätt symmetri. "
Kristallografisk symmetri ger reglerna som begränsar hur atomer kan ordnas i ett material, så fullt ut förstå symmetrin i en struktur är nödvändig för att korrekt beskriva den, Khalifah noterade.
Teamet hade genererat var och en av försöksstrukturerna med en specifik uppsättning symmetribegränsningar. Och även om det var mycket utmanande att bestämma den verkliga symmetrin för någon teststruktur efter dess optimering, en jämförelse av alla 2, 500 optimerade strukturer gjorde det möjligt för forskarna att avgöra vilka symmetrielement som behövdes för att korrekt beskriva den sanna strukturen för NVPF.
Möjligheten att jämföra resultat mellan många prövningar möjliggör en högre grad av förtroende för den slutliga lösningen och är en ytterligare fördel som den nya metoden som används i detta arbete har jämfört med traditionella tillvägagångssätt. Vidare, teoretiska beräkningar gjorda av LBNL -forskare John Dagdelen och Alex Ganose indikerade att den slutliga lösningen är stabil mot snedvridningar, bekräftar giltigheten av detta resultat.
Den lösta strukturen avslöjade att det finns mycket större mångfald i bindningen av natriumatomer än vad som tidigare hade erkänts.
"Från rumstemperaturdata, det visade sig vilseledande att alla natriumatomer var bundna till antingen sex eller sju angränsande atomer, "Sa Khalifah." Däremot, data från låg temperatur indikerade tydligt att vissa natriumatomer har så få som fyra grannar. Ett resultat av detta är att natriumatomerna med färre grannar är mycket mindre låsta på plats och därför förväntas ha lättare att röra sig genom strukturen - en egenskap som är avgörande för batterifunktion. "
Författarna tror att detta nya tillvägagångssätt bör vara i stort sett tillämpligt för att lösa de komplexa strukturer som vanligen förekommer i batterimaterial när joner avlägsnas under laddning. Detta är särskilt relevant i material som används i natrium- och kaliumjonbatterier, som utvecklas som billigare och rikligare alternativ till litiumjonbatterimaterial. Denna forskning bör därför spela en viktig roll när det gäller att frigöra potentialen hos jordmånga material som kan användas för att skala upp energilagringsmöjligheter för att möta samhällsbehov som lagring i nätskala.