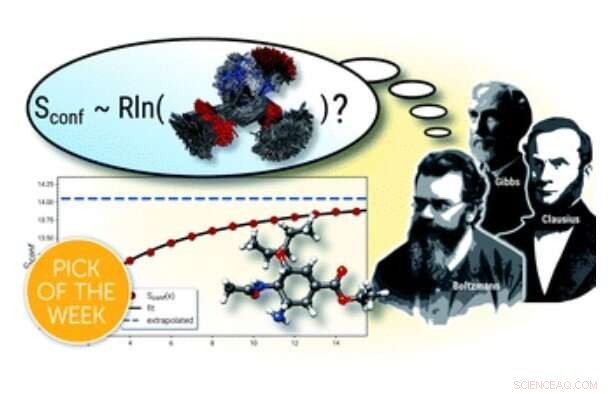
Grafiskt abstrakt. Kredit:Beräkning av absoluta molekylära entropier och värmekapacitet på ett enkelt sätt, Kemivetenskap (2021). DOI:10.1039/D1SC00621E
Kemister vid universitetet i Bonn utvecklade ett beräkningsverktyg för analys av konformationella entropier av flexibla molekyler. Deras metod möjliggör termodynamisk undersökning av komplicerade kemiska system genom kombination av moderna kvantkemiska och klassiska modeller. I ett framgångsrikt försök att förenkla, viktiga bidrag till entropin kan beräknas med minimalt användaringripande, även på vanliga stationära datorer. Resultaten publiceras i tidskriften Kemivetenskap och lyftes fram som artikeln "Veckans val".
Termen "entropi" introducerades 1865 av den tyske fysikern Rudolf Clausius, som senare arbetade och var rektor vid universitetet i Bonn. 2022 kommer att vara 200-årsdagen av hans födelsedag och vetenskapliga evenemang och firande planeras vid universitetet i Bonn. Entropi är en av de mest grundläggande termodynamiska egenskaperna hos materia och är vanligtvis förknippad med ett tillstånd av oordning eller osäkerhet. Med tiden har konceptet tagit fäste även inom statistisk mekanik, som pionjärer av kända fysiker Josiah Gibbs och Ludwig Boltzmann, och i informationsteori. I dag, entropi är ett aktivt forskningsområde inom många vetenskapliga områden, inklusive beräkningskemi.
För molekyler blir entropin viktig som en del av den temperaturberoende beskrivningen av det inre, så kallad fri energi, från vilka många egenskaper såsom kemiska jämvikter eller reaktionshastigheter härleds. I modern beräkningskemi, en molekyls entropi erhålls från energinivåer av atomvibrationer inom en molekylstruktur. Här, på grund av höga beräkningskostnader på kvantkemisk nivå flera teoretiska förenklingar, såsom den så kallade rigid-rotor harmonic-oscillator approximation, måste införas och beräkningar utförs för det mesta endast för en enda struktur. För flexibla molekyler leder detta till att ett viktigt bidrag som kallas konformationsentropin försummas, som beskriver den molekylära "störningen" från alla termiskt tillgängliga konformationer. Sådana flexibla fall är vanliga och viktiga för många läkemedel.
I ett nyligen försök att tillhandahålla korrekta temodynamiska beskrivningar av flexibla molekyler, Prof. Dr. Stefan Grimme och medarbetare från Mulliken Center for Theoretical Chemistry vid universitetet i Bonn utvecklade ett nytt beräkningsverktyg för beräkning av konformationella entropier. Medan matematiska formuleringar för beräkningar av den konformationella entropin har varit kända under ganska lång tid, ett huvudproblem är att hitta och utvärdera det enorma antalet möjliga strukturer som redan når miljarder för medelstora molekyler. Därav, en kärnkomponent i den nyligen introducerade och fritt tillgängliga programvaran är en effektiv algoritm för denna uppgift som fungerar med minimal användarinmatning, även på vanliga stationära datorer. För att uppnå den effektivitet som krävs, semiempriska kvantkemiska metoder tillämpades som också är utvecklade i Grimmes grupp, tillsammans med vanliga kvantmekaniska beräkningar. I artikeln visades det att proceduren kan behandla även stora och extremt flexibla system med oöverträffad noggrannhet för den molekylära entropin. Det är författarna som hoppas att det nya beräkningsprotokollet kan hjälpa till att erhålla korrekta termodynamiska data mer rutinmässigt och att det kommer att få en bred tillämpning inom beräkningskemi.
Stefan Grimmes forskargrupp arbetar med aktuella ämnen inom kvantkemi med fokus på beräkningseffektivitet och stora molekyler. Hans medarbetare Philipp Pracht håller för närvarande på att avsluta sin doktorsexamen. avhandling och är huvudförfattare till programmet CREST som används för de konformationella entropiberäkningarna. Denna forskning är publicerad med öppen tillgång i Kemivetenskap , Royal Society of Chemistrys referentgranskade flaggskeppstidskrift.