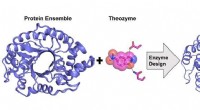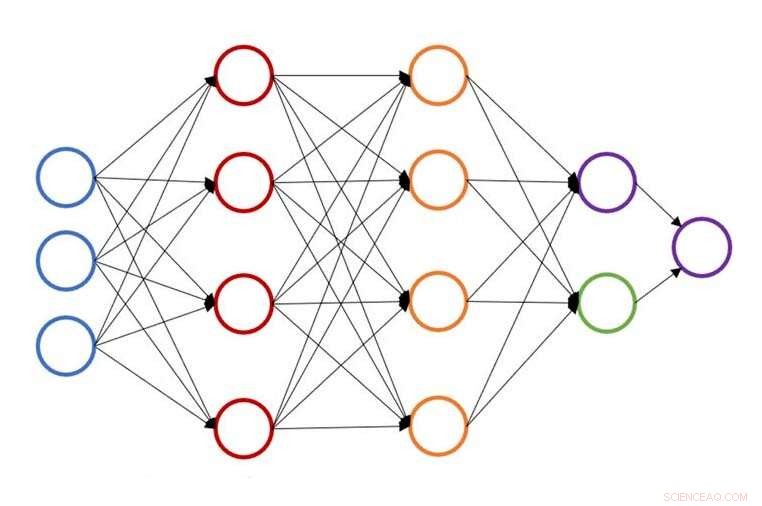
Illustration av en enda neural nätverksstruktur för atomenergiprognos. Upphovsman:John Kitchin, Carnegie Mellon University
Maskininlärning, en dataanalysmetod som används för att automatisera analytisk modellbyggnad, har omformat hur forskare och ingenjörer bedriver forskning. En gren av artificiell intelligens (AI) och datavetenskap, metoden bygger på ett stort antal algoritmer och breda datamängder för att identifiera mönster och fatta viktiga forskningsbeslut.
Tillämpningar av maskininlärningstekniker växer fram inom ytkatalys, möjliggör mer omfattande simuleringar av nanopartiklar, studier av segregation, strukturoptimering, on-the-fly inlärning av kraftfält, och screening med hög genomströmning. Dock, Att arbeta igenom stora mängder data kan ofta vara en lång och beräkningsmässigt dyr uppgift.
Geometrioptimering, ofta det hastighetsbegränsande steget i molekylära simuleringar, är en viktig del av beräkningsmaterial och ytvetenskap. Det gör det möjligt för forskare att hitta atomkonstruktioner och reaktionsvägar i grundtillstånd, egenskaper som används för att uppskatta de kinetiska och termodynamiska egenskaperna hos molekyl- och kristallstrukturer. Även om det är viktigt, processen kan vara relativt långsam, kräver ett stort antal beräkningar för att slutföra.
Vid Carnegie Mellon University, John Kitchin arbetar för att påskynda den processen genom att tillhandahålla en neural nätverksbaserad aktiv inlärningsmetod som accelererar geometrisk optimering för flera konfigurationer samtidigt. Den nya modellen sänker antalet densitetsfunktionella teorier (DFT) eller effektiva mediumteori (EMT) med 50 till 90 procent, låta forskare göra samma arbete på kortare tid eller mer arbete under samma tid.
"I vanliga fall, när vi gör geometrioptimering, vi börjar från början, "sa Kitchin." Beräkningarna drar sällan nytta av någonting vi visste tidigare. "
"Genom att lägga till en surrogatmodell i processen, vi har gjort det möjligt att förlita sig på tidigare beräkningar, snarare än att börja om från början varje gång. "
Studien illustrerar accelerationen i flera fallstudier, inklusive ytor med adsorbat, nakna metallytor, och ett elastiskt band för två reaktioner. I varje fall, Atomic Simulation Environment (ASE) -optimizer Python-paketet möjliggjorde färre DFT-beräkningar än standardmetoden.
ASE-optimizer Python-paketet har gjorts tillgängligt för andra ingenjörer och forskare för att göra användningen av neuralt nätverksensemble aktivt lärande för geometrioptimering enklare.