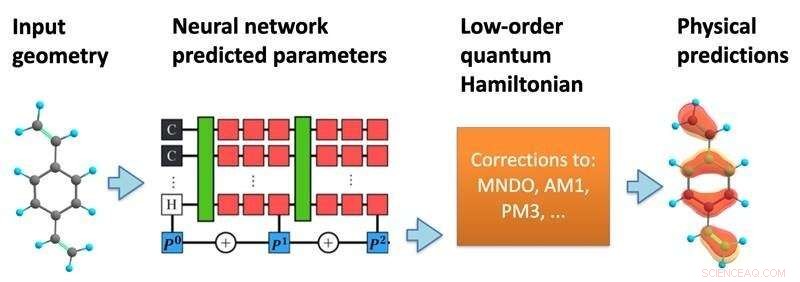
Modellens struktur. Ett neuralt nätverk bearbetar en molekylär geometri för att förutsäga en semi-empirisk kvant Hamiltonian, som sedan löses självkonsekvent för att förutsäga en mängd olika kemiska egenskaper. Kredit:Kipton Barros, Los Alamos National Laboratory.
I en ny studie, publicerad i Proceedings of the National Academy of Sciences , har forskare från Los Alamos National Laboratory föreslagit att införliva mer av kvantmekanikens matematik i strukturen av maskininlärningsförutsägelserna. Med hjälp av atomernas specifika positioner inom en molekyl förutsäger maskininlärningsmodellen en effektiv Hamiltonsk matris, som beskriver de olika möjliga elektroniska tillstånden tillsammans med deras associerade energier.
Jämfört med traditionella kvantkemi-simuleringar gör den maskininlärningsbaserade metoden förutsägelser till en mycket reducerad beräkningskostnad. Det möjliggör kvantitativt exakta förutsägelser om materialegenskaper, tillåter tolkningsbar insikt i naturen av kemisk bindning mellan atomer och kan användas för att förutsäga andra komplexa fenomen, såsom hur systemet kommer att reagera på störningar, såsom interaktioner mellan ljus och materia. Metoden ger också avsevärt förbättrad noggrannhet jämfört med traditionella maskininlärningsmodeller och visar framgång i överförbarhet, dvs modellens förmåga att göra förutsägelser som går långt utöver de data som låg till grund för dess träning.
Kvantmekanikens ekvationer ger en färdplan för att förutsäga kemikaliers egenskaper med utgångspunkt från grundläggande vetenskapliga teorier. Dessa ekvationer kan dock snabbt bli för dyra när det gäller datortid och kraft när de används för att förutsäga beteende i stora system. Maskininlärning erbjuder ett lovande tillvägagångssätt för att accelerera sådana storskaliga simuleringar. Användningen av maskininlärning för att förutsäga kemiska egenskaper har potentialen för stora tekniska framsteg, med tillämpningar från renare energi till snabbare läkemedelsdesign. Detta är ett mycket aktivt forskningsområde, men de flesta befintliga metoder använder enkla och heuristiska metoder för utformningen av maskininlärningsmodellerna.
I sin studie har forskarna visat att maskininlärningsmodeller kan efterlikna grundstrukturen i de grundläggande naturlagarna. Dessa lagar kan vara mycket svåra att simulera direkt. Maskininlärningsmetoden möjliggör förutsägelser som är lätta att beräkna och som är korrekta i ett brett spektrum av kemiska system.
Den förbättrade maskininlärningsmodellen kan snabbt och exakt förutsäga ett brett utbud av egenskaper hos molekyler. Dessa tillvägagångssätt ger mycket goda resultat på viktiga riktmärken inom beräkningskemi och visar hur metoder för djupinlärning kan fortsätta att förbättras genom att införliva mer data från experiment. Modellen kan också lyckas med utmanande uppgifter som att förutsäga upphetsad tillståndsdynamik – hur system beter sig med förhöjda energinivåer. Detta verktyg är en banbrytande förmåga för kvantkemi. Det kommer att tillåta forskare att bättre förstå reaktiviteten och exciterade tillstånd hos nya molekyler. + Utforska vidare