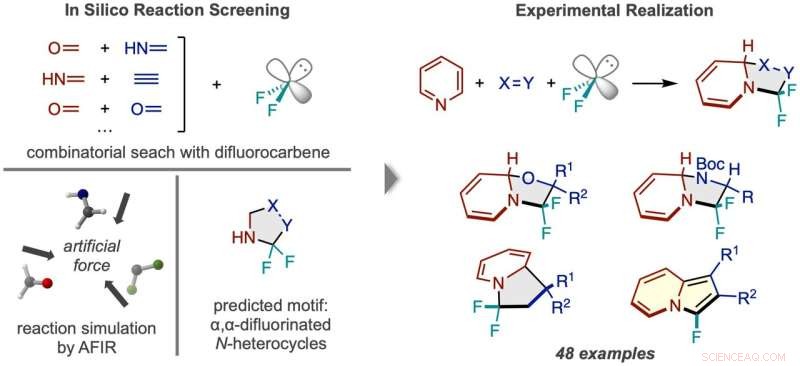
Arbetsflöde för reaktionsupptäckt via in silico screening. (Vänster) Reaktioner mellan difluorkarben och många par av små molekyler simulerades, vilket förutsäger en N-heterocykelprodukt fluorerad två gånger vid alfakolet. (Höger) Den framgångsrika reaktionsramen med pyridin och exempel på de typer av produktföreningar som erhålls. Kredit:Nature Synthesis (2022). DOI:10.1038/s44160-022-00128-y
Datorsimuleringar används oftast som en guide så att kemister mer effektivt kan räkna ut de exakta detaljerna i en allmän reaktionsidé de har i åtanke - ungefär som en kompass hjälper en utforskare effektivt att vägleda en destination på kartan. Men forskare vid ICReDD tog saker och ting ett stort steg längre och använde simuleringar för att producera den allmänna idén för en helt oanad reaktion, och effektivt använda beräkningar för att göra själva kartan. Med hjälp av designprincipen som föreslagits av beräkningsresultat, slog teamet till moderloden i labbet och utvecklade framgångsrikt en serie av 48 reaktioner som producerar föreningar som potentiellt är användbara för utveckling av nya läkemedel.
Närvaron och positionen av fluor i en molekyl påverkar ofta en molekyls farmakologiska aktivitet. Forskare vid ICReDD har använt kvantkemiska beräkningar för att upptäcka en reaktion som selektivt adderar två fluoratomer till en svåråtkomlig position på en N-heterocykel - molekyler med en kolringstruktur där minst ett kol i ringen är ersatt med kväve . Förmågan att fästa fluoratomer till det tidigare svåråtkomliga "alfakolet" - kolet omedelbart bredvid kvävet i ringstrukturen - kan leda till utvecklingen av en mängd nya läkemedel.
Innan de utförde experiment i labbet, kastade forskarna ett brett nät, och testade beräkningsmässigt livsdugligheten hos många 3-komponentsreaktioner med hjälp av den artificiella kraftinducerade reaktionsmetoden (AFIR). De simulerade reaktionen av en difluorkarbenmolekyl, som verkar vid källan till fluoratomer, med olika par av små molekyler med en dubbel- eller trippelbindning. Dessa simuleringar visade att ett antal ringbildande reaktioner borde vara genomförbara.
Forskare försökte en av de lovande reaktionerna som föreslogs av initiala beräkningsresultat men lyckades inte. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis . + Utforska vidare