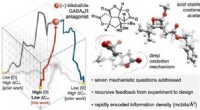
Genererade strukturer och reaktionsvägnätverk för de förutsagda reaktionsstegen för Strecker-reaktionen (a) och Passerini-reaktionen (b). Vita pilar visar flerstegsvägen som motsvarar den kända reaktionsmekanismen. Kredit:Satoshi Maeda
Forskare har övervunnit beräkningsbegränsningar för att förutsäga utgångsmaterialen för flerstegsreaktioner med endast information om målproduktmolekylen. Deras studie är publicerad i JACS Au .
Har du någonsin bara fångat slutet på ett TV-program och undrat hur historien utvecklades till det slutet? På liknande sätt har kemister ofta en önskad molekyl i åtanke och undrar vilken typ av reaktion som skulle kunna ge den. Forskare i Maeda-gruppen vid Institutet för kemisk reaktionsdesign och upptäckt (ICReDD) och Hokkaido-universitetet utvecklade en metod som kan förutsäga "berättelsen" (d.v.s. utgångsmaterialen och reaktionsvägarna) för kemiska reaktioner i flera steg genom att endast använda information om "slutet" (d.v.s. produktmolekylerna).
Att förutsäga receptet för en målproduktmolekyl, utan någon annan kunskap än själva molekylen, skulle vara ett kraftfullt verktyg för att påskynda upptäckten av nya reaktioner. Maeda-gruppen utvecklade tidigare en beräkningsmetod som på detta sätt lyckades förutsäga enstegsreaktioner. Utvidgning till flerstegsreaktioner leder dock till en dramatisk ökning av antalet möjliga reaktionsvägar - det som kallas kombinatorisk explosion. Denna kraftiga ökning av komplexiteten resulterar i oöverkomligt höga beräkningskostnader.
För att övervinna denna begränsning utvecklade forskare en algoritm som minskar antalet vägar som behöver utforskas genom att kassera mindre livskraftiga vägar vid varje steg i reaktionen. Efter att ha beräknat alla möjliga vägar för ett steg bakåt i reaktionen, utvärderar en kinetisk analysmetod hur väl varje väg producerar målmolekylen. Reaktionsvägar som inte ger målmolekylen över en förinställd tröskelprocentandel anses inte vara tillräckligt signifikanta och utforskas inte vidare.
Denna cykel av att utforska, utvärdera och kassera reaktionsvägar upprepas för varje steg bakåt i en flerstegsreaktion och mildrar den kombinatoriska explosion som normalt skulle inträffa, vilket gör flerstegsreaktioner mer genomförbara att beräkna. Tidigare metoder var begränsade till enstegsreaktioner, medan den här nya metoden kunde förutsäga reaktioner som involverade mer än sex steg, vilket markerade ett stort hopp i förmåga.
Som ett proof-of-concept-test testade forskare metoden på två välkända flerstegsreaktioner, Strecker- och Passerini-reaktionerna. Tusentals startmaterialkandidater föreslogs för varje reaktion, vilka filtrerades till de mest lovande kandidaterna baserat på stabilitet och produktutbyte. Av avgörande betydelse var bland de föreslagna kandidaterna de välkända utgångsmaterialen för varje reaktion, vilket bekräftar teknikens förmåga att identifiera experimentellt livskraftiga utgångsmaterial från bara målproduktmolekylen.
Även om ytterligare arbete krävs för att göra det möjligt att förutsäga ännu större och mer komplexa system, förutser forskarna att detta genombrott i hanteringen av flerstegsprocesser kommer att påskynda upptäckten av nya kemiska reaktioner.
"Detta arbete ger ett unikt tillvägagångssätt, eftersom det är första gången att utföra omvända förutsägelser av flerstegsreaktioner med hjälp av kvantkemiska beräkningar är möjligt utan att använda någon kunskap eller data om reaktionen", säger professor Satoshi Maeda. "Vi förväntar oss att denna teknik kommer att möjliggöra upptäckten av helt oanade kemiska transformationer, i vilket fall det finns lite kunskap eller experimentella data att använda." + Utforska vidare