Forskare från Cleveland Clinic och IBM har nyligen publicerat resultat i Journal of Chemical Theory and Computation som skulle kunna lägga grunden för att tillämpa kvantberäkningsmetoder för att förutsäga proteinstruktur.
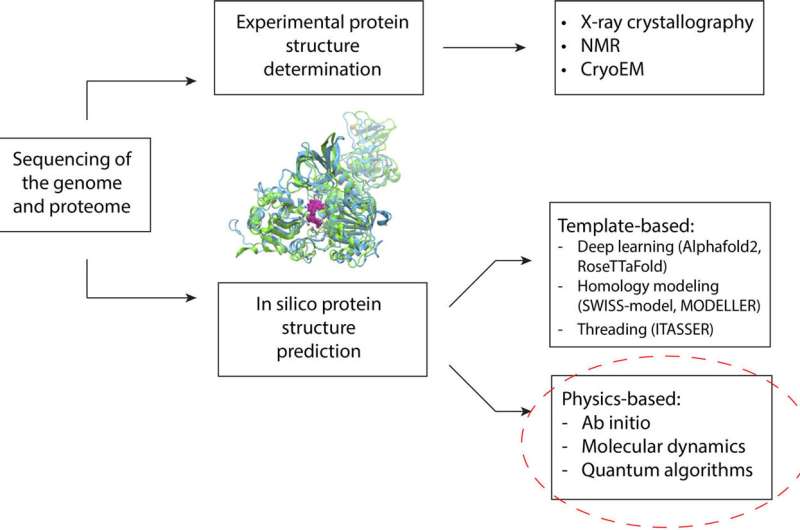
I decennier har forskare utnyttjat beräkningsmetoder för att förutsäga proteinstrukturer. Ett protein viker sig till en struktur som bestämmer hur det fungerar och binder till andra molekyler i kroppen. Dessa strukturer bestämmer många aspekter av människors hälsa och sjukdomar.
Genom att noggrant förutsäga ett proteins struktur kan forskare bättre förstå hur sjukdomar sprids och därmed hur man utvecklar effektiva terapier. Cleveland Clinic postdoktor Bryan Raubenolt, Ph.D. och IBM-forskaren Hakan Doga, Ph.D. ledde ett team för att upptäcka hur kvantberäkning kan förbättra nuvarande metoder.
Under de senaste åren har maskininlärningstekniker gjort betydande framsteg i förutsägelse av proteinstruktur. Dessa metoder är beroende av träningsdata (en databas med experimentellt bestämda proteinstrukturer) för att göra förutsägelser. Detta innebär att de är begränsade av hur många proteiner de har lärt sig att känna igen. Detta kan leda till lägre nivåer av noggrannhet när programmen/algoritmerna stöter på ett protein som är muterat eller mycket annorlunda än de som de tränades på, vilket är vanligt med genetiska störningar.
Den alternativa metoden är att simulera fysiken för proteinveckning. Simuleringar gör det möjligt för forskare att titta på ett givet proteins olika möjliga former och hitta den mest stabila. Den mest stabila formen är avgörande för läkemedelsdesign.
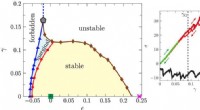
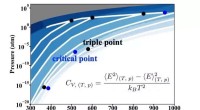
Utmaningen är att dessa simuleringar är nästan omöjliga på en klassisk dator, bortom en viss proteinstorlek. På ett sätt är att öka storleken på målproteinet jämförbart med att öka dimensionerna på en Rubiks kub. För ett litet protein med 100 aminosyror skulle en klassisk dator behöva den tid som är lika med universums ålder för att uttömmande söka igenom alla möjliga resultat, säger Dr. Raubenolt.
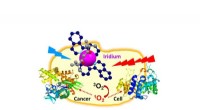
För att hjälpa till att övervinna dessa begränsningar använde forskargruppen en blandning av kvantberäkningsmetoder och klassiska beräkningsmetoder. Detta ramverk skulle kunna tillåta kvantalgoritmer att ta itu med de områden som är utmanande för toppmodern klassisk datoranvändning, inklusive proteinstorlek, inneboende störning, mutationer och fysiken involverad i proteinveckning. Ramverket validerades genom att exakt förutsäga veckningen av ett litet fragment av ett Zika-virusprotein på en kvantdator, jämfört med toppmoderna klassiska metoder.
Det kvantklassiska hybridramverkets initiala resultat överträffade både en klassisk fysikbaserad metod och AlphaFold2. Även om det senare är designat för att fungera bäst med större proteiner, visar det ändå detta ramverks förmåga att skapa korrekta modeller utan att direkt förlita sig på betydande träningsdata.
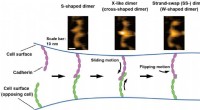
Forskarna använde en kvantalgoritm för att först modellera den lägsta energikonformationen för fragmentets ryggrad, vilket vanligtvis är det mest beräkningskrävande steget i beräkningen. Klassiska tillvägagångssätt användes sedan för att omvandla resultaten från kvantdatorn, rekonstruera proteinet med dess sidokedjor och utföra slutlig förfining av strukturen med klassiska molekylära mekaniska kraftfält.
Projektet visar ett av sätten som problem kan dekonstrueras till delar, med kvantberäkningsmetoder som adresserar vissa delar och klassiska beräkningar andra, för ökad noggrannhet.
"En av de mest unika sakerna med det här projektet är antalet inblandade discipliner", säger Dr Raubenolt. "Vårt teams expertis sträcker sig från beräkningsbiologi och kemi, strukturbiologi, mjukvaru- och automationsteknik, till experimentell atom- och kärnfysik, matematik, och naturligtvis, kvantberäkning och algoritmdesign. Det krävdes kunskap från vart och ett av dessa områden för att skapa en beräkningsramverk som kan efterlikna en av de viktigaste processerna för mänskligt liv."
Teamets kombination av klassiska och kvantberäkningsmetoder är ett viktigt steg för att förbättra vår förståelse av proteinstrukturer och hur de påverkar vår förmåga att behandla och förebygga sjukdomar. Teamet planerar att fortsätta utveckla och optimera kvantalgoritmer som kan förutsäga strukturen hos större och mer sofistikerade proteiner.
"Detta arbete är ett viktigt steg framåt för att utforska var kvantberäkningskapacitet kan visa styrkor i förutsägelse av proteinstruktur", säger Dr. Doga. "Vårt mål är att designa kvantalgoritmer som kan hitta hur man förutsäger proteinstrukturer så realistiskt som möjligt."
Mer information: Hakan Doga et al, A Perspective on Protein Structure Prediction Using Quantum Computers, Journal of Chemical Theory and Computation (2024). DOI:10.1021/acs.jctc.4c00067
Tillhandahålls av Cleveland Clinic