I en artikel som nyligen publicerades i Nature Communications , HUN-REN-ELTE Protein Modeling Research Group (Institute of Chemistry) har lagt grunden för en matematisk metod som möjliggör datorstödd jämförelse av proteiners tredimensionella strukturer. Metoden är unik genom att medan de alternativ som finns tillgängliga hittills endast tagit hänsyn till atomernas position, inkluderar den nya tekniken, kallad LoCoHD (Local Composition Hellinger Distance), även atomernas kemiska information.
Proteiner är molekylära maskiner som utför processer som är nödvändiga för att celler ska fungera, fungerar som molekylära switchar, transkriberar information från DNA, transporterar små och stora molekyler och reglerar metabolismrelaterade kemiska reaktioner. Men för att allt detta ska lyckas måste proteinet i fråga ha rätt rumslig konformation, det vill säga sitt eget, korrekta 3D-arrangemang.
Flera experimentella metoder (röntgenkristallografi, kärnmagnetisk resonansspektroskopi, kryo-elektronmikroskopi) finns tillgängliga för att bestämma arrangemanget av atomer i ett protein, och under de senaste decennierna har proteinforskare upptäckt formen på nästan 220 000 proteiner. Dessa resultat kräver alltmer utveckling av beräkningsmetoder som kan analysera dessa arrangemang.
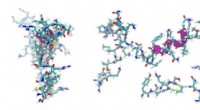
En sådan metod är algoritmen som kallas LoCoHD, utvecklad av Zsolt Fazekas, en Ph.D. kandidat vid ELTE Hevesy György School of Chemistry och forskare i Dr. András Perczels forskargrupp. Algoritmen jämför lokala miljöer kring aminosyror i proteiner baserat på deras kemiska natur (t.ex. elementär sammansättning, laddning, hydrofobicitet, etc.).
Metoden avgör på en enkel skala från 0 till 1 hur olika strukturerna i fråga är från varandra. Värden nära 0 tyder på en hög likhet mellan atomarrangemang och kemiska egenskaper, medan värden nära 1 indikerar att de proteiner som jämförs kan ha mycket olika egenskaper. Det resulterande numeriska värdet (en så kallad metrik) kan alltså användas för att få ny information om det studerade systemet.
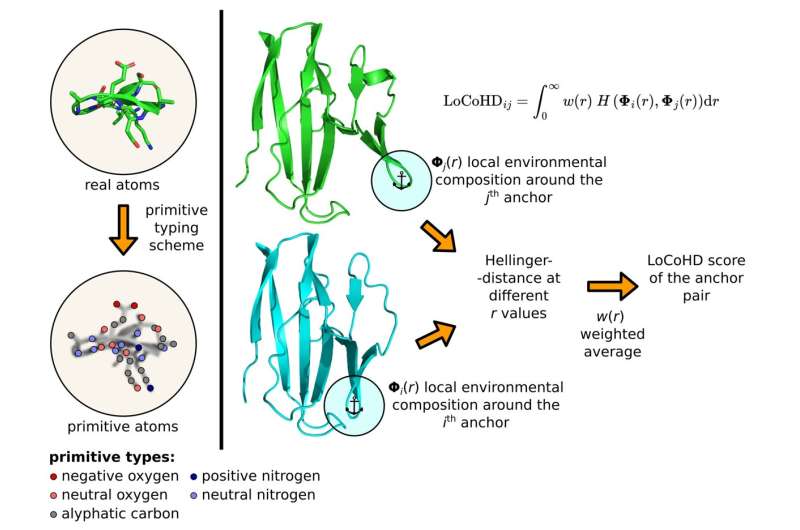
Algoritmen använder ett flerstegsprotokoll för att generera numret som representerar de strukturella skillnaderna. I det första steget omvandlar den verkliga atomer i proteinet till så kallade primitiva atomer. Dessa kan representeras som praktiskt taget märkta positioner vars etiketter berättar om den ursprungliga atomens kemiska natur.
Så till exempel kan en primitiv atom vara ett "positivt laddat kväve", ett "negativt laddat syre", ett "neutralt laddat syre", ett "aromatiskt kol" etc. Etiketterna genereras enligt en så kallad primitiv typningsschema, som berättar för oss i tabellform hur man omvandlar verkliga atomer till primitiva atomer. Användaren kan fritt specificera denna tabell och fixera metodens kemiska upplösning.
Det andra steget är att bestämma referenspunkterna för jämförelsen genom att välja en undergrupp av primitiva atomer. Dessa utvalda speciella primitiva atomer kallas för ankaratomer. För varje valt ankaratompar utför algoritmen ett jämförelsesteg, vars resultat ger det olikhetsmått vi vill ha. Dessa siffror kan användas på lokal nivå, eller så kan de sättas i medeltal till en enda deskriptor som karakteriserar hela proteinet.
I studien framhöll forskarna att metoden även kan användas i de tvååriga CASP-tävlingarna (Critical Assessment of Protein Structure Prediction), som är en välkänd tävling inom proteinforskningsområdet. Under detta evenemang använder de tävlande olika algoritmer för att modellera formen på proteiner med ännu opublicerade strukturer. CASP-domare använder ett antal strukturjämförelsemetoder för att utvärdera utmanarna, men ingen av dessa tar hänsyn till kemin i de lokala aminosyramiljöerna.
Med hjälp av data från 2020 CASP14-tävlingen har forskarna nu utfört jämförande analys av flera modellerade proteiner, inklusive de strukturer som förutspåtts av den artificiella intelligensbaserade AlphaFold2-metoden. Bland dessa lyfte de fram analysen av ett protein från SARS-CoV-2-viruset som heter ORF8. I de modellerade strukturerna för detta protein identifierades aminosyramiljöer som skiljer sig signifikant i sina interaktionsmönster från de miljöer som finns i den experimentella strukturen.
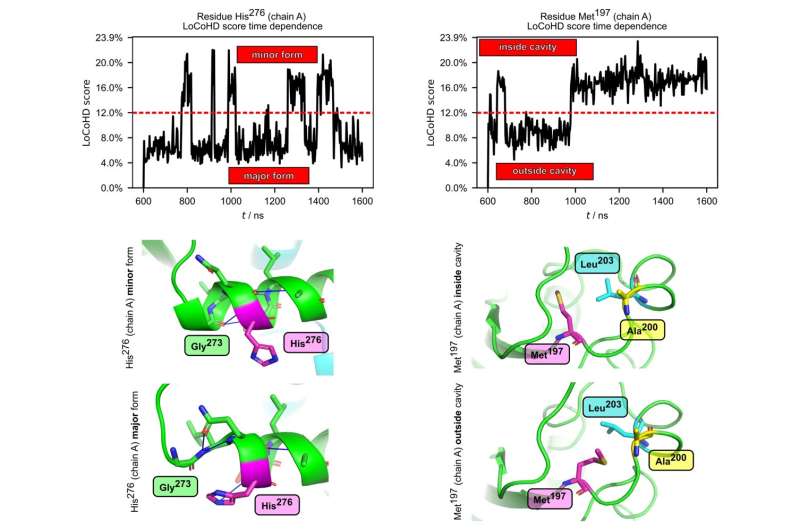
Förutom att studera statiska strukturer testade forskarna även om metoden är lämplig för att analysera proteiners inre rörelse. De använde simuleringar som kan reproducera molekylära rörelser och data som extraherats från strukturella ensembler. Ett av systemen som studerades var podocinproteinet, som utför vitala funktioner i njuren och vars mutationer kan orsaka svåra, ofta dödliga tillstånd.
LoCoHD-metoden användes för att identifiera aminosyror i proteinet som genomgår stora kemiska-miljöförändringar under podocins rörelse, vilket kan påverka både dess struktur och funktion. På liknande sätt har LoCoHD-metoden använts framgångsrikt i studien av kapsidproteinet HIV-1, där en aminosyra som är avgörande för bildandet av virushöljet har identifierats.
Dessa resultat är inte bara forskningskuriosa, utan genom att studera proteinstrukturer mer effektivt kan vi komma närmare att bättre förstå de patogener som orsakar allvarliga sjukdomar och att utveckla effektiva läkemedel och terapier.
Mer information: Zsolt Fazekas et al, LoCoHD:ett mått för att jämföra lokala miljöer av proteiner, Nature Communications (2024). DOI:10.1038/s41467-024-48225-0
Journalinformation: Nature Communications
Tillhandahålls av Eötvös Loránd University