En ny datorprocess utvecklad av kemister vid ETH Zürich gör det möjligt att snabbt och enkelt generera aktiva läkemedelsingredienser baserat på ett proteins tredimensionella yta. Den nya processen, som beskrivs i Nature Communications , skulle kunna revolutionera läkemedelsforskningen.
"Det är ett verkligt genombrott för läkemedelsupptäckten", säger Gisbert Schneider, professor vid ETH Zürichs institution för kemi och tillämpad biovetenskap. Tillsammans med sin tidigare doktorand Kenneth Atz har han utvecklat en algoritm som använder artificiell intelligens (AI) för att designa nya aktiva läkemedelsingredienser.
För alla proteiner med en känd tredimensionell form, genererar algoritmen ritningarna för potentiella läkemedelsmolekyler som ökar eller hämmar proteinets aktivitet. Kemister kan sedan syntetisera och testa dessa molekyler i laboratoriet.
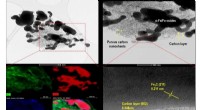
Allt algoritmen behöver är ett proteins tredimensionella ytstruktur. Utifrån det designar den molekyler som binder specifikt till proteinet enligt lås-och-nyckel-principen så att de kan interagera med det.
Den nya metoden bygger på kemisters decennier långa ansträngningar att belysa proteiners tredimensionella struktur och att använda datorer för att söka efter lämpliga potentiella läkemedelsmolekyler. Hittills har det ofta inneburit mödosamt manuellt arbete och i många fall har sökningen gett molekyler som varit mycket svåra eller omöjliga att syntetisera. Om forskare överhuvudtaget använt AI i denna process under de senaste åren så var det främst för att förbättra befintliga molekyler.
Nu, utan mänsklig inblandning, kan en generativ AI utveckla läkemedelsmolekyler från grunden som matchar en proteinstruktur. Denna banbrytande nya process säkerställer redan från början att molekylerna kan syntetiseras kemiskt. Dessutom föreslår algoritmen endast molekyler som interagerar med det specificerade proteinet på önskad plats och knappast alls med några andra proteiner.
"Detta betyder att när vi designar en läkemedelsmolekyl kan vi vara säkra på att den har så få biverkningar som möjligt", säger Atz.
För att skapa algoritmen tränade forskarna en AI-modell med information från hundratusentals kända interaktioner mellan kemiska molekyler och motsvarande tredimensionella proteinstrukturer.
Tillsammans med forskare från läkemedelsföretaget Roche och andra samarbetspartners testade ETH-teamet den nya processen och visade vad den kan.
Forskarna sökte efter molekyler som interagerar med proteiner i PPAR-klassen - proteiner som reglerar socker- och fettsyrametabolismen i kroppen. Flera diabetesläkemedel som används idag ökar aktiviteten av PPAR, vilket gör att cellerna tar upp mer socker från blodet och blodsockernivån sjunker.
Genast designade AI nya molekyler som också ökar aktiviteten hos PPAR, som de läkemedel som för närvarande finns tillgängliga, men utan en lång upptäcktsprocess. Efter att ETH-forskarna hade producerat dessa molekyler i labbet, utsatte kollegor på Roche dem för en mängd olika tester. Dessa visade att de nya ämnena verkligen är stabila och giftfria redan från början.
Forskarna söker nu inte längre dessa molekyler i syfte att föra ut läkemedel baserade på dem på marknaden. Istället var syftet med molekylerna att utsätta den nya AI-processen för ett första rigoröst test.
Schneider säger dock att algoritmen redan används för liknande studier vid ETH Zürich och inom industrin. Ett av dessa är ett projekt med Barnsjukhuset Zürich för behandling av medulloblastom, de vanligaste maligna hjärntumörerna hos barn. Dessutom har forskarna publicerat algoritmen och dess mjukvara så att forskare över hela världen nu kan använda dem för sina egna projekt.
"Vårt arbete har gjort en värld av proteiner tillgänglig för generativ AI i läkemedelsforskning", säger Schneider. "Den nya algoritmen har enorm potential." Detta gäller särskilt för alla medicinskt relevanta proteiner i människokroppen som inte interagerar med några kända kemiska föreningar.
Mer information: Kenneth Atz et al, Prospective de novo drug design with deep interactome learning, Nature Communications (2024). DOI:10.1038/s41467-024-47613-w
Journalinformation: Nature Communications
Tillhandahålls av ETH Zürich