Kemister utvecklar och optimerar ofta nya kemiska reaktioner med hjälp av så kallade modellsystem, det vill säga enkla, lättillgängliga substrat. De använder sedan upp till runt 100 andra substrat som exempel för att visa att reaktionen fungerar. Denna demonstration av mångsidig användbarhet kallas "omfattning" på teknisk jargong.
Ett subjektivt urval av substrat resulterar emellertid ofta i en förvrängd bild av tillämpningsområdet för den nyutvecklade reaktionen. Det är ofta oklart om det kan användas för att syntetisera en önskad produkt. För att ta itu med detta problem föreslår ett team under ledning av kemisten prof Frank Glorius från universitetet i Münster (Tyskland) en datorstödd, fördomsfri metod för att välja modellsubstrat för att utvärdera nya kemiska reaktioner.
Urvalet av substrat baseras på komplexiteten och strukturella egenskaperna hos verkliga farmaceutiska föreningar. "Vår metod syftar till att förbättra kvaliteten och informationsinnehållet i kemiska reaktionsdata i framtiden och att täppa till kunskapsluckor", förklarar Glorius.
En djupare förståelse för nya reaktioner sänker barriärerna för deras tillämpning i både ett akademiskt och industriellt sammanhang. Tillgången på högkvalitativ, opartisk data underlättar också avsevärt användningen av maskininlärning och banar väg för en mer omfattande användning av datan. Arbetet har publicerats i tidskriften ACS Central Science .
Enligt teamets författare är försök att standardisera och objektivera utvecklingen och utvärderingen av kemiska reaktioner fortfarande ganska nya och relativt ovanliga. "Vi skulle vilja inleda en 'omtänkande process' med vår publicering. Istället för att göra så många experiment som möjligt, som ofta är partiska eller har ett förutsägbart resultat, bör fokus ligga på att få fram bästa möjliga data om nya kemiska reaktioner." säger första författaren Debanjan Rana.
Andra forskare har också försökt utvärdera kemiska reaktioner utifrån "bättre" utvalda substrat. Detta arbete var dock begränsat till speciella fall – antingen till bestämt utvalda strukturer med farmaceutisk relevans eller till strukturer speciellt anpassade för en enstaka reaktion, som måste beräknas och väljas i en komplex process.
I motsats till det tidigare arbetet tar metoden som presenterades av Münster-teamet hänsyn till hela strukturen hos en molekyl, vilket gör den universell tillämpbar på alla kemiska reaktioner.
Niklas Hölter, en av tidningens författare i Münster, förklarar tankeprocessen bakom studien:"Omfattning är av central betydelse i alla publikationer om kemisk syntes. Men kemister är ofta partiska i sitt val av substratföreningar att testa.
"De väljer till exempel substrat som är strukturellt mycket enkla, mycket lik modellsubstratet eller helt enkelt bara tillgängliga i laboratoriet ("selektionsbias"). Ofta nämner de inte misslyckade reaktioner alls i sin publikation för att måla en bättre bild ('reporting bias')."
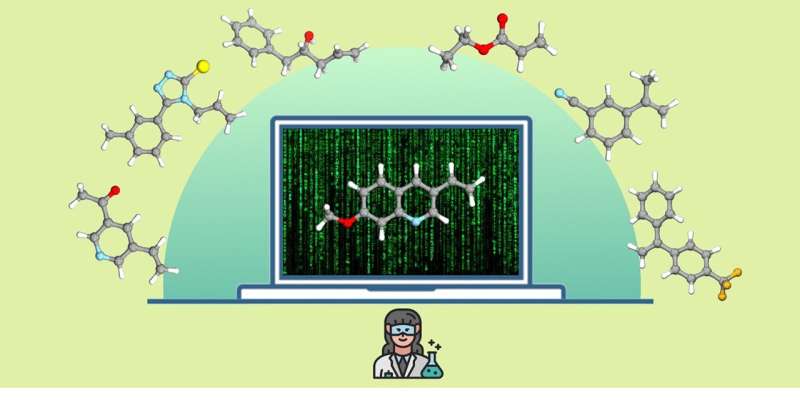
Vid syntetisering av nya kemiska föreningar, såsom aktiva ingredienser eller material, måste kemister välja den mest lämpliga metoden för att framställa målföreningen från ett stort antal kända kemiska reaktioner och metoder. För att göra detta tar de hänsyn till flera faktorer, såsom utbytet av den önskade produkten samt miljö- och säkerhetsaspekter. Utvecklingen av nya, mångsidiga kemiska reaktioner fortsätter därför att vara ett fokus för aktuell kemisk forskning.
Metoden som utvecklats av teamet vid universitetet i Münster använde molekylära fingeravtryck för att överföra alla godkända aktiva läkemedelsingredienser till en digital kod. Med hjälp av oövervakade maskininlärning och klustringsmetoder skapade de en modell som delar upp detta "utrymme" av aktiva farmaceutiska ingredienser i kemiskt meningsfulla regioner baserade på molekylära strukturer.
För att utvärdera en ny kemisk reaktion kan tusentals potentiella testsubstrat projiceras in i samma utrymme med hjälp av maskininlärningsmodellen. Ett testsubstrat väljs automatiskt från mitten av var och en av de tidigare identifierade regionerna för att täcka hela utrymmet utan förspänning.
Mer information: Debanjan Rana et al, Standardizing Substrate Selection:A Strategy toward Unbiased Evaluation of Reaction Generality, ACS Central Science (2024). DOI:10.1021/acscentsci.3c01638
Journalinformation: ACS Central Science
Tillhandahålls av University of Münster