En forskargrupp från Skoltech introducerade en ny metod som drar fördel av maskininlärning för att studera egenskaperna hos polykristaller, kompositer och flerfassystem. Den uppnådde hög noggrannhet, nästan lika bra som kvantmekaniska metoder, som bara är tillämpliga på material med mindre än några hundra atomer.
Den nya metoden drar också nytta av aktivt lärande på lokala atommiljöer. Uppsatsen publiceras i Advanced Theory and Simulations journal.
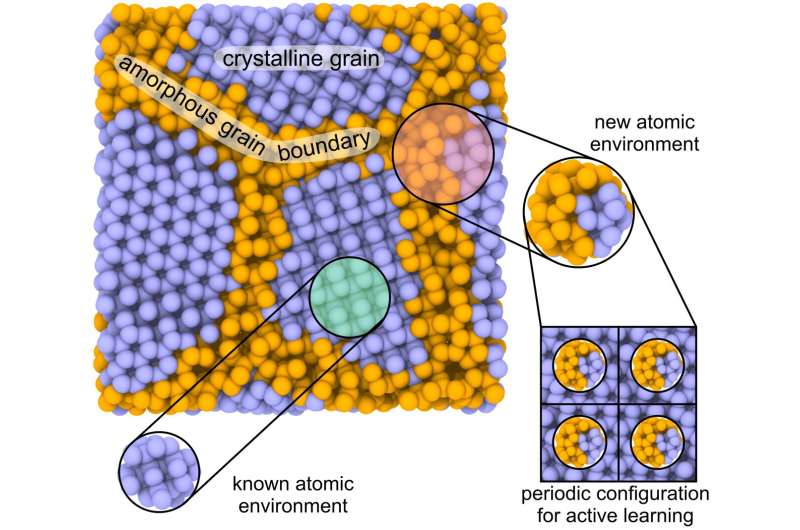
"Många industriella material syntetiseras som polykristaller eller flerfassystem. De innehåller både enkristall och amorfa komponenter mellan enkristallkorn. Det stora antalet atomer gör det svårt att beräkna egenskaperna hos dessa system med moderna kvantmekaniska metoder. Densitet funktionell teori kan bara tillämpas på material med några hundra atomer."
"För att komma till rätta med problemet använder vi en maskininlärningsmetod baserad på Moment Tensor Potentials (MTP). Dessa potentialer har också utvecklats vid Skoltech under ledning av professor Alexander Shapeev", kommenterade Faridun Jalolov, den ledande författaren av studien och en Skoltech Ph.D. student på Materialvetenskapsprogrammet.
Jämfört med andra lösningar ser författarna potentialen med den nya metoden i aktivt lärande på lokala atomära miljöer. Vid beräkning av en stor struktur med många hundratusentals atomer identifierar MTP:n vilken atom som gör ett misstag i beräkningarna, eller beräknas felaktigt. Anledningen till detta kan vara den begränsade träningsdatauppsättningen, som förhindrar att alla möjliga systemkonfigurationer beaktas.
En lokal miljö för denna atom "klipps ut" och dess energi beräknas med hjälp av kvantmekanik. Efteråt läggs data tillbaka till träningsuppsättningen för vidare inlärning. Allt eftersom inlärningen fortskrider, fortsätter beräkningarna tills de stöter på en annan konfiguration som måste inkluderas i utbildningsprocessen. Andra kända maskininlärningspotentialer kan inte läras in på små lokala delar av stora strukturer, vilket begränsar deras tillämpbarhet och noggrannhet.
"Som ett exempel studerade vi de mekaniska egenskaperna hos diamantpolykristaller, som är de hårdaste naturligt förekommande materialen och ofta används inom industrin — till exempel vid tillverkning av borrutrustning för oljekällor. Resultaten visar att de mekaniska egenskaperna hos dessa polykristallina diamanter beror på på kornstorleken – ju större korn, desto mer liknar egenskaperna egenskaperna hos en enkristalldiamant", fortsatte Jalolov.
Författarna påpekade att detta tillvägagångssätt kommer att göra det möjligt att studera de mekaniska egenskaperna hos icke-enkristallina material som vanligtvis syntetiseras och används i experiment, samt att genomföra omfattande studier av polykristallina och kompositmaterial och erhålla data så nära experimentella resultat som möjligt.
"Vid faktisk användning används ofta material som inte är perfekta kristaller på grund av deras oförmåga att perfekta kristaller uppfyller kraven för en specifik utrustning helt."
"Ett bra exempel på detta är volframkarbid och kobolt. Genom att tillsätta kobolt till volframkarbid blir materialet mer sprickbeständigt, vilket gör det så värdefullt i applikationer. Den nya metoden kommer att göra det möjligt för oss att undersöka orsakerna och sätten att förändra det mekaniska egenskaper hos dessa flerfassystem på atomnivå", säger Alexander Kvashnin, forskningschef och professor vid Energy Transition Center.
Mer information: Faridun N. Jalolov et al, Mechanical Properties of Single and Polycrystalline Solids from Machine Learning, Advanced Theory and Simulations (2024). DOI:10.1002/adts.202301171
Tillhandahålls av Skolkovo Institute of Science and Technology