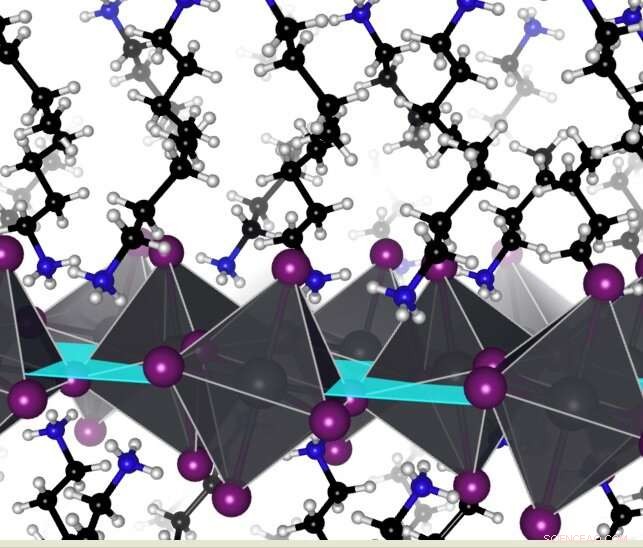
Hybridperovskiter är som molekylär baklava med alternerande metallbaserade oorganiska skikt och kolbaserade organiska skikt. Interaktioner i det organiska skiktet kan justera förvrängningar i den oorganiska komponenten och öka effektiviteten hos solceller gjorda av dessa material. Kredit:Arvin Kakekhani
På en enda dag träffar tillräckligt med solljus jorden för att driva världen under ett helt år - det vill säga om vi kan hitta ett sätt att fånga den energin billigt och effektivt. Medan kostnaden för solenergi har minskat dramatiskt, är nuvarande kiselbaserade solceller dyra och energikrävande att tillverka, vilket får forskare att leta efter alternativ.
Perovskite solceller är en främsta utmanare för nästa generation av denna förnybara energi. Dessa syntetiska material är billigare och kräver mindre energi att producera men hamnar efter många kiselbaserade celler när det gäller deras stabilitet och effektivitet. Nu, en artikel publicerad i Nature Communications från grupperna av University of Pennsylvanias Andrew M. Rappe och Yueh-Lin Loo från Princeton University ger insikt i hur molekylär sammansättning av vissa perovskiter kan påverka deras effektivitet och erbjuder en väg framåt till bättre solceller med hjälp av en enkel metrisk.
"Världen behöver för närvarande mer effektiva och kostnadseffektiva solceller, och 3D-hybrid perovskite PV har tagit världen med storm", säger Rappe, professor vid Penns avdelning för kemi som också leder Penns VIPER-program. "Men de är oåterkalleligt skadade av vatten, vilket är en showstopper för praktiska tillämpningar. Att infoga organiska molekylära plan mellan 2D-hybridperovskitplan är ett lovande system för att tillhandahålla effektiva, billiga och robusta solceller."
I den här studien undersökte forskarna en viss klass av perovskiter som kallas 2D-hybridperovskiter. Jämfört med perovskiter gjorda av 3D-kristaller tenderar dessa att vara mer stabila, byggda som molekylär baklava med omväxlande lager av metall- och kolbaserade molekyler. Det metallbaserade skiktet, som kallas det oorganiska skiktet, interagerar med ljus för att producera elektricitet och är mest effektivt när dess atomer anpassas ordentligt. Det kolbaserade, eller organiska, skiktet är sammansatt av positivt laddade molekyler som balanserar det negativt laddade oorganiska skiktet.
Till en början förberedde Princeton-teamet en uppsättning 2D-perovskiter med olika organiska molekyler, och studerade hur dessa molekyler påverkade det oorganiska lagrets inriktning och solcellseffektiviteten. I synnerhet tittade de på en klass av korta, flexibla organiska molekyler, var och en med en positiv laddning i ena änden. De märkte att typen av molekyl påverkade strukturen och energieffektiviteten hos solcellerna men visste inte exakt varför eller hur. De behövde en atomistisk insikt för att komplettera de experimentella fynden och hypoteserna. Detta skulle hjälpa till att förklara systemets höga prestanda.
Så de nådde ut till Rappe och Arvin Kakekhani, då postdoc i Rappe-gruppen, experter på att använda datorer för att modellera kemiska interaktioner. "[Princetonforskarna] är mycket intelligenta experimentalister och hade stor insikt på experimentnivå", säger Kakekhani. "Men de behövde kunskap och insikt på atomär, molekylär nivå." Det är just den typen av arbete som Rappe-labbet utmärker sig i, efter att tidigare ha samarbetat med Loo-gruppen för att modellera andra perovskitmaterial i samband med att rationalisera deras mekaniska egenskaper.
Från de nuvarande kvantmekaniska beräkningarna och laddningsmodelleringsarbetet fann Kakekhani och Rappe att molekylerna i det organiska skiktet kunde interagera med varandra, rada upp sig i par eller i sicksack mellan perovskiternas metallbaserade skikt.
När de bildade dessa par eller sicksack, interagerade de organiska molekylerna mindre med det metallbaserade lagret, vilket gav lagret utrymme att rikta in sig ordentligt och förbättrade prestandan hos de resulterande solcellerna. Ju lättare de organiska molekylerna kunde paras ihop och komma ur vägen för det oorganiska lagret, desto bättre effektivitet har den resulterande solcellen.
Bara denna observation erbjöd insikt i hur man gör bättre perovskiter. Men Kakekhani undrade om han kunde hitta ett sätt att fånga detta fenomen i ett enkelt värde som beskrev interaktionen mellan de organiska och oorganiska lagren. Efter att ha testat olika modeller landade han på en som beskrev hur långt bort interaktionerna i det organiska lagret drog den positiva laddningen från det oorganiska lagret. Sedan testade han det för att se om det kan förutsäga hur väl det oorganiska lagret skulle anpassa sig och hur bra solcellerna skulle kunna prestera.
I stället för att anpassa en modell med hjälp av data från experimentet, valde han att bygga den enbart med hjälp av den matematiska och fysiska förståelsen av hur kemikalier interagerar. Detta är känt som materialmodellering med de första principerna.
Dessa typer av modeller kämpar ofta för att exakt replikera verkliga resultat, eftersom de kan vara för enkla, bara med tanke på en liten delmängd av möjliga fenomen involverade i ett komplext experiment. First-principles modellering blir mer kraftfull när den kan ge fysisk insikt och förbättra förståelsen för hur man reducerar ett komplext problem till ett enklare utan att det skadar modellens trohet.
I det här fallet förutspådde Kakekhani de verkliga trenderna med förvånansvärt hög trohet. I matematiska termer ger hans modell en bestämningskoefficient på>0,95, nästan en perfekt linjär korrelation. "Jag hade aldrig sett en så perfekt överensstämmelse mellan modeller med första principer och komplexa experimentella observerbara objekt tidigare", säger Kakekhani. "Att koppla en modell som sitter i en dator och inte vet något om experimentet till verklig materia med alla möjliga defekter och strukturer i större skala - det var verkligen överraskande."
Eftersom detta mått bara behöver en dator för att förutsäga solcellsprestanda, kan det tillåta forskare att välja vilka molekyler som kan fungera bäst i perovskiter innan de går in i labbet, vilket hjälper forskare att begränsa sina ansträngningar till endast de mest lovande kandidaterna. "Det finns bokstavligen miljontals molekyler som folk skulle kunna prova. Men det är inte så lätt att göra miljontals solceller", säger Rappe. "Detta ger människor en enkel poängregel, där de kan analysera om en molekyl de överväger sannolikt kommer att förbättra solcellens produktivitet."
I framtiden säger Rappe att dessa insikter också kan hjälpa till med perovskite-lysdioder. Om dessa perovskiter kan förvandla ljus till energi effektivt, borde de kunna göra något liknande när de förvandlar energi till ljus. Grupperna planerar att se om samma modell gäller för olika oorganiska lager och ett bredare utbud av organiska molekyler, eller om andra faktorer behöver beaktas för att exakt modellera perovskiten.
Men för närvarande använder modellen ett värde för att förutsäga prestandan hos en komplex solcell, och modellens enkelhet är dess styrka, säger Kakekhani. "Enkelhet skapar insikt, och den insikten kan verkligen skapa stora framsteg inom vetenskapen eftersom den går in i den olinjära kreativa delen av din hjärna. Den stannar där och den hjälper dig att komma på alla möjliga intuitioner." + Utforska vidare