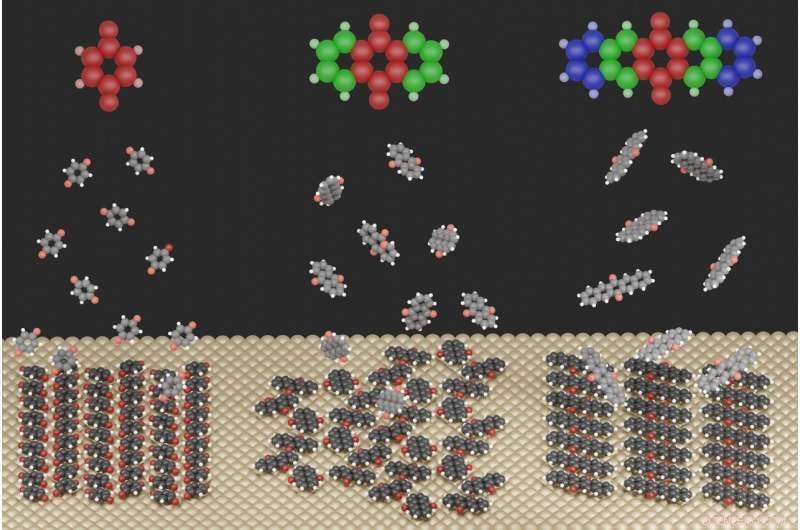
Illustrationen visar de starkt olika ytstrukturer som bildas för de tre studerade molekylerna när de adsorberas på en metallyta. Kredit:Jeindl—TU Graz
Med hjälp av maskininlärningsmetoder, forskare vid TU Graz kan förutsäga strukturbildningen av funktionaliserade molekyler vid gränssnitten mellan hybridmaterial. Nu har man också lyckats se bakom drivkrafterna i denna strukturbildning.
Produktionen av nanomaterial involverar självmonteringsprocesser av funktionaliserade (organiska) molekyler på oorganiska ytor. Denna kombination av organiska och oorganiska komponenter är avgörande för tillämpningar inom organisk elektronik och andra områden av nanoteknik.
Tills nu, vissa önskade ytegenskaper uppnåddes ofta på prov-och-fel-basis. Molekyler modifierades kemiskt tills det bästa resultatet för den önskade ytegenskapen hittades. Dock, processerna som styr självsammansättningen av molekyler vid gränssnitt är så komplexa att små molekylära förändringar kan leda till helt andra motiv. Fysiker från TU Graz förklarar denna oväntade strukturbildning i en studie publicerad i den berömda tidskriften ACS Nano .
För det här syftet, forskarna studerade kinoidföreningar på en silveryta. Första författaren Andreas Jeindl från Institute of Solid State Physics förklarar:"Naivt, man kan förvänta sig att molekyler med lite olika storlekar men samma funktionalisering bildar liknande motiv. I slående kontrast, vår gemensamma teoretiska och experimentella studie visar att kinoner kan bilda olika strukturer. Trots konstanta initiala förhållanden, bildandet av dessa strukturer kan inte förutsägas och planeras utan detaljerad kunskap om de relevanta interaktionerna."
Tre motsatta drivkrafter
Forskarna i Graz, tillsammans med ett team från FSU Jena, har nu börjat bryta ner denna oförutsägbarhet. De fann att strukturbildningen är resultatet av en avvägning mellan tre motsatta drivkrafter:Interaktionen mellan molekyler och metallen försöker tvinga alla molekyler till samma orientering, medan interaktionen mellan molekyler ibland gynnar olika orienteringar. Molekylernas geometriska former fungerar då som en tredje faktor, förhindra eller endast delvis tillåta vissa interaktioner.
Baserat på det här, de kunde etablera en designprincip med vilken strukturerna som bildas vid gränssnitten, och därefter deras egenskaper, kan förutsägas - åtminstone för en första klass av molekyler. En viktig roll spelas av en sökalgoritm (SAMPLE) baserad på maskininlärning. Jeindl utvecklar:"Vi kunde visa i denna publikation att de strukturer som förutspås av vår algoritm är i utmärkt överensstämmelse med experimentella karakteriseringar av organisk-oorganiska gränssnitt - både i hur molekylerna orienterar sig på ytan och i hur motiven upprepas på ytan. yta. Dessutom vår analys, för första gången, möjliggjorde en detaljerad och kvantitativ nedbrytning av drivkrafterna, inte bara av de experimentellt formade strukturerna, men de facto av alla tänkbara strukturer. Detta är en viktig titt bakom kulisserna för strukturbildning."
Gränssnittsegenskaper med modulära byggstenar
Det icke-intuitiva samspelet mellan liknande viktiga interaktionsmekanismer är fortfarande en utmaning för utformningen av funktionella gränssnitt. Med en detaljerad undersökning av alla drivkrafter, dock, fysikerna vid TU Graz kan ändå ta fram en designprincip för självmontering av funktionaliserade molekyler för en given klass av molekyler. När det väl finns tillräckligt med analyser för olika klasser av molekyler, de rätta molekylerna för de önskade gränssnittsegenskaperna kan enkelt sättas ihop på datorn från modulära byggstenar.