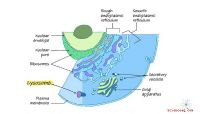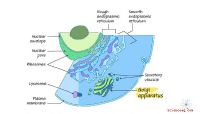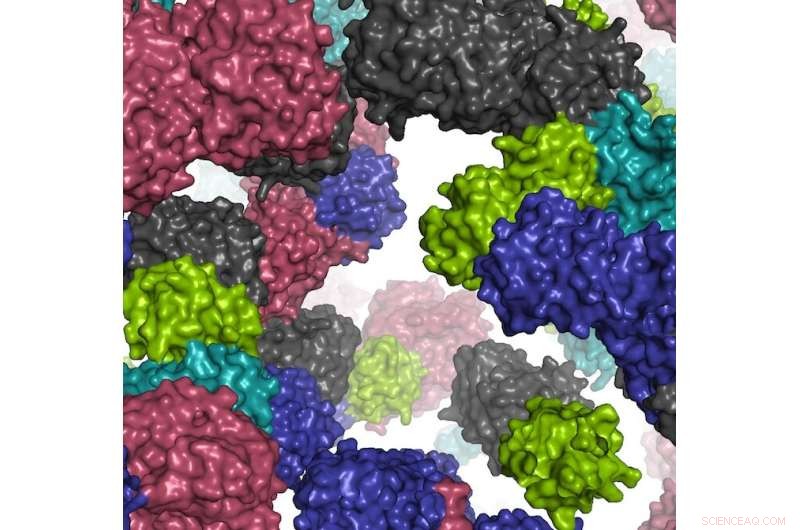
Ett fragment av den simulerade cellmiljön. Kredit:Ilya Vakser
En milstolperapport från University of Kansas som visas denna vecka i Proceedings of the National Academy of Sciences föreslår en ny teknik för att modellera molekylärt liv med datorer.
Enligt huvudförfattaren Ilya Vakser, chef för Computational Biology Program och Center for Computational Biology och professor i molekylär biovetenskap vid KU, är undersökningen av datormodellering av livsprocesser ett stort steg mot att skapa en fungerande simulering av en levande cell i atomär upplösning . Framsteg lovar nya insikter i en cells grundläggande biologi, samt snabbare och mer exakt behandling av mänskliga sjukdomar.
"Det är ungefär tiotals eller hundratusentals gånger snabbare än de befintliga atomupplösningsteknikerna," sa Vakser. "Detta ger oöverträffade möjligheter att karakterisera fysiologiska mekanismer som nu ligger långt bortom räckhåll för beräkningsmodellering, att få insikter i cellulära mekanismer och att använda denna kunskap för att förbättra vår förmåga att behandla sjukdomar."
Fram till nu har ett stort hinder för att modellera celler via dator varit hur man närmar sig proteiner och deras interaktioner som ligger i hjärtat av cellulära processer. Hittills har etablerade tekniker för att modellera proteininteraktioner varit beroende av antingen "proteindockning" eller "molekylär simulering."
Enligt utredarna har båda tillvägagångssätten fördelar och nackdelar. Även om proteindockningsalgoritmer är bra för att ta prover på rumsliga koordinater, tar de inte hänsyn till "tidskoordinaten" eller dynamiken i proteininteraktioner. Däremot modellerar molekylära simuleringar dynamiken väl, men dessa simuleringar är för långsamma eller för lågupplösta.
"Vår proof-of-concept-studie överbryggar de två modelleringsmetoderna och utvecklar ett tillvägagångssätt som kan nå oöverträffade simuleringstidsskalor med en atomupplösning", skrev författarna.
Vaksers medarbetare på tidningen var Sergei Grudinin vid universitetet i Grenoble Alpes i Frankrike; Eric Deeds från University of California-Los Angeles; KU-doktoranden Nathan Jenkins och Petras Kundrotas, biträdande forskningsprofessor vid KU:s Computational Biology Program.
Efter att ha konceptualiserat hur man bäst kombinerar fördelarna med de två proteinmodelleringsmetoderna, utvecklade och kodade teamet en algoritm för att driva den nya simuleringen.
"Den svåraste utmaningen var att utveckla algoritmen som adekvat återspeglar den enkla grundidén för tillvägagångssättet," sa Vakser.
Men när de väl fick det genombrottet kunde de börja validera den nya proceduren.
"Paradigmet var lätt - ett slag av klarhet," sa Vakser.
"De befintliga simuleringsmetoderna tillbringar större delen av beräkningstiden på att resa i områden med låg sannolikhet eller hög energi i systemet. Vi vet alla var dessa områden finns. Istället var tanken att prova, eller resa, bara i de höga områdena. -sannolikhet, lågenergiområden, och att hoppa över de låga sannolikhetsområdena genom att uppskatta övergångshastigheterna mellan högsannolikhetstillstånden. Paradigmet är lika gammalt som själva biomolekylära modelleringen och har använts flitigt sedan modelleringstidens gryning decennier sedan."
Men Vakser sa fram till hans teams nya artikel, att tillvägagångssättet inte hade tillämpats på kinetiken för proteininteraktioner i cellulär miljö, i fokus för deras studie.
"Eftersom det finns mycket färre tillstånd med hög sannolikhet än de med låg sannolikhet, gav det oss en enorm vinst i beräkningshastigheten - tiotals till hundratusentals gånger," sa Vakser. "Detta gjordes utan uppenbar förlust av noggrannhet. Man kan argumentera för att noggrannheten uppnåddes, eftersom simuleringsprotokollet är baserat på "dockningstekniker", som är speciellt utformade för att karakterisera proteinsammansättningar."
KU-forskaren sa att hans cellsimuleringsmetod skulle kunna användas för att undersöka människors hälsa och behandla sjukdomar med en ny nivå av precision.
"Tillvägagångssättet kan användas för att studera molekylära vägar som ligger bakom sjukdomsmekanismer," sade Vakser. "Den kan användas för att fastställa skadliga effekter av genetiska mutationer genom de förändrade mönstren av proteinassociationer – genetiska mutationer orsakar förändringar i proteiners struktur, vilket i sin tur påverkar proteinassociationen. Eller så kan den användas för att identifiera mål för läkemedelsdesign av detektera kritiska element i proteinassociationsmönster."
Enligt Vakser erbjuder den nya simuleringstekniken många lovande forskningsvägar att utforska framöver.
"Bland dem är att anpassa tillvägagångssättet för proteininteraktioner med nukleinsyror, RNA och DNA," sa han. "Vi skulle också vilja ta hänsyn till flexibiliteten hos molekylära former, korrelera med det snabbt växande spektrumet av experimentella studier av den cellulära miljön och tillämpa proceduren på en modell av en verklig cell - med dess faktiska molekylära komponenter packade tillsammans." + Utforska vidare