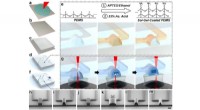
Lysosomer är membranbundna organeller som innehåller hydrolytiska enzymer som bryter ner en mängd olika molekyler, inklusive proteiner, lipider och kolhydrater. Lysosomer är viktiga för att upprätthålla cellulär homeostas, men de kan också vara farliga om de spricker och släpper ut innehållet i cytoplasman. För att förhindra detta har celler ett antal mekanismer på plats för att säkerställa att skadade lysosomer snabbt identifieras och tas bort.
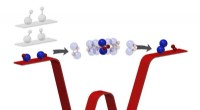
En av de viktigaste mekanismerna för att välja ut skadade lysosomer för clearance är processen för ubiquitination. Ubiquitin är ett litet protein som kan fästas till andra proteiner för att markera dem för nedbrytning. När en lysosom är skadad, rekryteras ubiquitinligaser till skadeplatsen och de fäster ubiquitinmolekyler till det lysosomala membranet. Detta markerar lysosomen för igenkänning av autofagireceptorer, som är proteiner som binder till ubiquitin och riktar sig mot lysosomen för leverans till autofagosomen, en dubbelmembranvesikel som smälter samman med lysosomen för att leverera dess innehåll för nedbrytning.
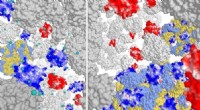
I en nyligen genomförd studie använde forskare vid University of California, San Francisco, molekylära taggar för att spåra ödet för skadade lysosomer i celler. De fann att ubiquitinering inte är den enda mekanismen som kan markera skadade lysosomer för clearance. De identifierade också ett antal andra molekylära taggar som kan användas för att välja ut skadade lysosomer, inklusive:
* HMGB1: HMGB1 är ett protein som frigörs från kärnan när celler är stressade. HMGB1 kan binda till det lysosomala membranet och rekrytera autofagireceptorer.
* LC3: LC3 är ett protein som är involverat i autofagosomerbildning. LC3 kan också binda till det lysosomala membranet och rekrytera autofagireceptorer.
* p62: p62 är ett protein som är involverat i aggregeringen av skadade proteiner. p62 kan binda till det lysosomala membranet och rekrytera autofagireceptorer.
Forskarna fann att dessa molekylära taggar arbetar tillsammans för att säkerställa att skadade lysosomer snabbt identifieras och tas bort från celler. Denna process är väsentlig för att upprätthålla cellulär homeostas och förhindra utvecklingen av lysosomala lagringssjukdomar.
Konsekvenser för lysosomala lagringssjukdomar
Lysosomala lagringssjukdomar är en grupp genetiska störningar som orsakas av mutationer i gener som kodar för proteiner som är involverade i lysosomal funktion. Dessa mutationer leder till ackumulering av osmält material i lysosomer, vilket kan skada celler och vävnader. Lysosomala lagringssjukdomar är ofta dödliga och det finns för närvarande inget botemedel.
Forskningen som beskrivs ovan ger nya insikter om de mekanismer som celler använder för att välja och ta bort skadade lysosomer. Denna kunskap kan leda till utvecklingen av nya terapier för lysosomala lagringssjukdomar. Det kan till exempel vara möjligt att utveckla läkemedel som hämmar ubiquitination av lysosomer eller som blockerar bindningen av autofagireceptorer till lysosomer. Dessa läkemedel kan hjälpa till att förhindra ansamling av osmält material i lysosomer och bromsa utvecklingen av lysosomala lagringssjukdomar.