Metanhydrat hämtat från havsbotten utanför Oregons kust, USA. Kredit:Wikimedia Commons
I en tidning som publicerades denna vecka i PNAS , forskare vid universitetet i Amsterdams Van 't Hoff Institute for Molecular Sciences och Amsterdam Centre for Multiscale Modeling ger atomistisk insikt i bildandet av metanhydrater. På grundval av molekylära dynamiksimuleringar förklarar de hur urval mellan konkurrerande metanhydratpolymorfer sker, och hur detta kan generaliseras till andra hydrater och molekylär kristallbildning.
Metanhydrat är isliknande fasta ämnen som förekommer rikligt, bland annat vid havsbotten. Det uppskattas att energimängden som lagras i metanhydrat är dubbelt så mycket energi som lagras i konventionella fossila bränslen. På samma gång, bildandet av hydrater är ett problem för petroleumindustrin eftersom de kan täppa till oljeledningar, orsakar flödesproblem. Metanhydrater finns också i permafrosten i arktiska regioner. Upptining av permafrosten till följd av stigande globala temperaturer kan leda till utsläpp av stora mängder metan, som är en kraftfull växthusgas.
Metade molekyler
I ett metanhydrat, på molekylär nivå är metan inneslutet i ett vätebundet vattennätverk. Medan metangas är hydrofob under omgivningsförhållanden, vid låga temperaturer och höga tryck kan en blandning av vatten och metangas spontant nukleera till hydrater.
Över åren, intresset för att förstå hydraternas bildningsmekanism har ökat enormt. Särskilt deras bildning under naturliga förhållanden är dåligt förstådd. Förstå processen för homogen kärnbildning, och hur detta leder till olika metanhydratpolymorfer, kan leda till förbättrad kontroll av kristallisation, samt insikt i polymorfval i allmänhet.
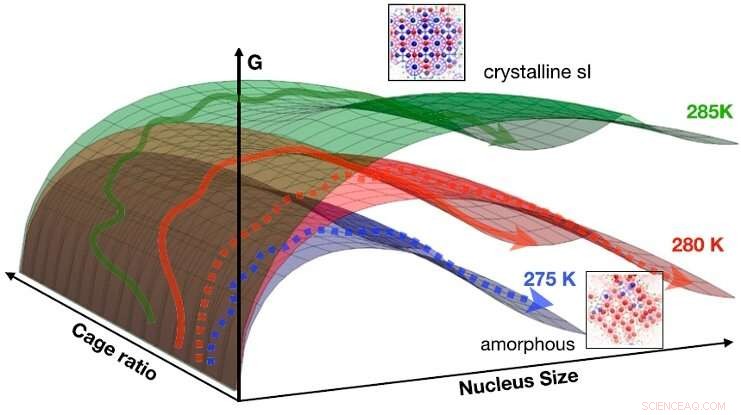
Fynden kan sammanfattas i en idealiserad CNT-fri energiyta som en funktion av storlek och burförhållande för 275 K (blå), 280 K (röd), och 285 K (grön). Pilarna indikerar schematiskt vägar som går från flytande till fast fas (streckade pilar:till amorf fas; heldragna pilar:till kristallin fas). Vid låga temperaturer (t.ex. 275 K), den fria energibarriären för att kärnbilda det amorfa fasta ämnet är lägst, trenden vänds vid högre temperatur (t.ex. 285 K), där provade vägar mestadels hamnar i den kristallina fasen. Vid 280 K är båda mekanismerna tillgängliga. Kredit:HIMS/PNAS
Ny simuleringsmetod
Eftersom experimentell forskning om bildandet av olika metanhydratpolymorfer lider av begränsad upplösning, forskarna i Amsterdam under ledning av professor Peter Bolhuis använde simuleringar av molekylär dynamik för att ge sådan insikt.
Att tillämpa en direkt molekylär dynamiksimulering är inte särskilt effektivt, eftersom kärnbildning vid måttlig underkylning är en mycket sällsynt händelse, på grund av närvaron en mycket hög energibarriär. En sådan simulering skulle kräva beräkningstider bortom universums ålder. Dock, eftersom kärnbildningshändelsen i sig, medan det är sällsynt, sker mycket snabbt (på en mikrosekund tidsskala), forskarna kunde skapa en stor samling av molekylära dynamikbanor som visar dessa snabba händelser. Efterföljande detaljerad analys av dessa banor visade hur urval mellan konkurrerande amorfa och kristallina polymorfbildningsmekanismer sker. Deras PNAS-papper belyser inte bara bildandet av metanhydrater, men också på andra klatratföreningar och molekylär kristallbildning i allmänhet.