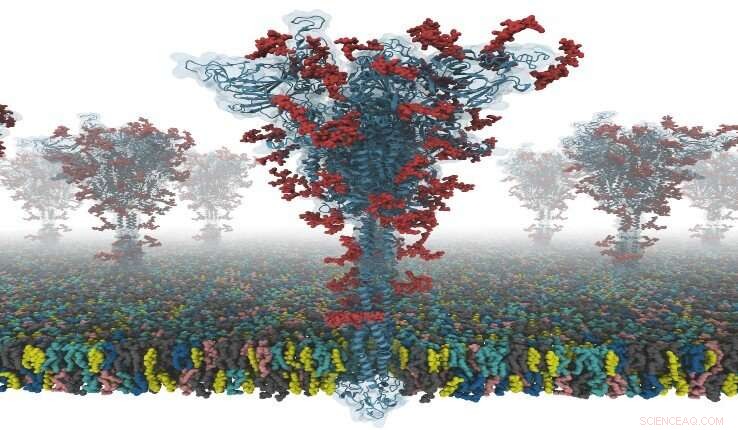
En modell av ett S-protein. Upphovsman:Dr Yeolkyo Choi/Lehigh
Viruset SARS coronavirus 2 (SARS-CoV-2) är den kända orsaken till coronavirus-sjukdomen 2019 (COVID-19). "Spiken" eller S -proteinet underlättar viralt inträde i värdceller.
Nu är en grupp forskare från Seoul National University i Sydkorea, University of Cambridge i Storbritannien, och Lehigh University i USA, har arbetat tillsammans för att producera de första open-source all-atom-modellerna av ett S-protein i full längd. Forskarna säger att detta är av särskild vikt eftersom S -proteinet spelar en central roll vid virusinträde i celler, vilket gör det till ett huvudmål för utveckling av vaccin och antivirala läkemedel.
Detaljerna finns i ett papper, "Utveckla en fullt glykosylerad SARS-CoV-2 Spike Protein-modell i full längd i ett viralt membran" publicerad just online i Journal of Physical Chemistry B .
Denna videodemo illustrerar hur man bygger detta membransystem från deras SARS-CoV-2 S proteinmodeller. Modellbyggnadsprogrammet är öppen åtkomst och kan hittas från CHARMM-GUI:s hemsida genom att klicka på länken COVID-19 Arkiv, eller genom att klicka på arkivlänken i rubriken, sedan länken COVID-19 Proteins i vänstra sidofältet.
Utvecklad av Wonpil Im, professor vid institutionen för biologiska vetenskaper och bioteknik vid Lehigh University, CHARMM-GUI (GUI =grafiskt användargränssnitt) är ett program som simulerar komplexa biomolekylära system helt enkelt, exakt och snabbt. Jag beskriver det som ett "beräkningsmikroskop" som gör det möjligt för forskare att förstå interaktioner på molekylär nivå som inte kan observeras på något annat sätt. Mer information om CHARMM-GUI finns i den här videon.
"Våra modeller är de första helt glykosylerade SARS-CoV-2 spikproteinmodellerna (S) som är tillgängliga för andra forskare, "säger Im." Jag hade turen att samarbeta med Dr. Chaok Seok från Seoul National University i Korea och Dr Tristan Croll från University of Cambridge i Storbritannien. Vårt team tillbringade dagar och nätter med att bygga dessa modeller mycket noga från det kända kryo- EM -strukturportioner. Modellering var mycket utmanande eftersom det fanns många regioner där enkel modellering inte gav högkvalitativa modeller. "
Forskare kan använda modellerna för att genomföra innovativ och ny simuleringsforskning för förebyggande och behandling av COVID-19, enligt Im.
S-proteinstrukturen bestämdes med cryo-EM med RBD upp (PDB ID:6VSB), och med RBD ned (PDB ID:6VXX). Men, denna modell har många saknade rester. Så, de modellerade först de saknade aminosyraresterna, och sedan andra saknade domäner. Dessutom, de modellerade alla potentiella glykaner (eller kolhydrater) fästa vid S -proteinet. Dessa glykaner förhindrar antikroppsigenkänning, vilket gör det svårt att utveckla ett vaccin. De byggde också ett viralt membransystem av ett S -protein för simulering av molekylär dynamik.