
Shantenu Jha, ordförande för Brookhaven Labs Center for Datadriven Discovery, och hans team från Rutgers University och University College London designade en mjukvaruram för att exakt och snabbt beräkna hur starkt läkemedelskandidater binder till sina målproteiner. Ramverket syftar till att lösa det verkliga problemet med läkemedelsdesign – för närvarande en lång och dyr process – och kan ha en inverkan på personlig medicin. Kredit:Brookhaven National Laboratory
Lösningar på många verkliga vetenskapliga och tekniska problem – från att förbättra vädermodeller och designa nya energimaterial till att förstå hur universum bildades – kräver applikationer som kan skalas till en mycket stor storlek och hög prestanda. Varje år, genom sin International Scalable Computing Challenge (SCALE), Institute of Electrical and Electronics Engineers (IEEE) erkänner ett projekt som främjar applikationsutveckling och stödjande infrastruktur för att möjliggöra storskaliga, högpresterande beräkningar som behövs för att lösa sådana problem.
Årets vinnare, "Möjliggör avvägning mellan noggrannhet och beräkningskostnad:Adaptiva algoritmer för att minska tiden till klinisk insikt, " är resultatet av ett samarbete mellan kemister och beräknings- och datavetare vid US Department of Energy's (DOE) Brookhaven National Laboratory, Rutgers University, och University College London. Teammedlemmarna hedrades vid det 18:e IEEE/Association for Computing Machinery (ACM) International Symposium on Cluster, Cloud and Grid Computing hölls i Washington, DC, från 1 till 4 maj.
"Vi utvecklade en numerisk beräkningsmetodik för att noggrant och snabbt utvärdera effektiviteten hos olika läkemedelskandidater, " sa teammedlemmen Shantenu Jha, ordförande för Center for Datadriven Discovery, del av Brookhaven Labs Computational Science Initiative. "Även om vi ännu inte har tillämpat denna metod för att designa ett nytt läkemedel, vi visade att det kunde fungera i den stora skala som är involverad i läkemedelsupptäcktsprocessen."
Att hitta droger är ungefär som att designa en nyckel för att passa ett lås. För att ett läkemedel ska vara effektivt vid behandling av en viss sjukdom, det måste binda hårt till en molekyl – vanligtvis ett protein – som är förknippad med den sjukdomen. Först då kan läkemedlet aktivera eller hämma målmolekylens funktion. Forskare kan screena 10, 000 eller fler molekylära föreningar innan man hittar någon som har den önskade biologiska aktiviteten. Men dessa "bly" föreningar saknar ofta styrkan, selektivitet, eller stabilitet som behövs för att bli ett läkemedel. Genom att modifiera den kemiska strukturen hos dessa ledningar, forskare kan designa föreningar med lämpliga läkemedelsliknande egenskaper. De designade läkemedelskandidaterna går sedan längs utvecklingspipelinen till det prekliniska teststadiet. Av dessa kandidater, endast en liten del går in i den kliniska prövningsfasen, och endast ett blir ett godkänt läkemedel för patientanvändning. Att ta ut ett nytt läkemedel på marknaden kan ta ett decennium eller längre och kosta miljarder dollar.
Att övervinna flaskhalsar i läkemedelsdesign genom beräkningsvetenskap
De senaste framstegen inom teknik och kunskap har resulterat i en ny era av läkemedelsupptäckt – en som avsevärt kan minska tiden och kostnaden för läkemedelsutvecklingsprocessen. Förbättringar i vår förståelse av biologiska molekylers 3D-kristallstrukturer och ökningar i datorkraft gör det möjligt att använda beräkningsmetoder för att förutsäga interaktioner mellan läkemedel och mål.
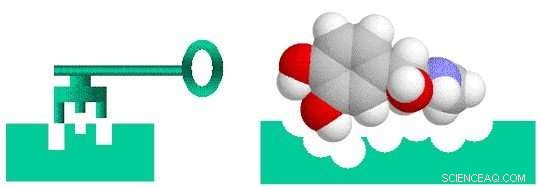
Läkemedelsupptäckt är ett lås-och-nyckel-problem där drogen (nyckeln) specifikt måste passa det biologiska målet (låset). Kredit:Brookhaven National Laboratory
Särskilt, en datorsimuleringsteknik som kallas molekylär dynamik har visat lovande när det gäller att exakt förutsäga styrkan med vilken läkemedelsmolekyler binder till sina mål (bindningsaffinitet). Molekylär dynamik simulerar hur atomer och molekyler rör sig när de interagerar i sin miljö. I fallet med upptäckt av droger, simuleringarna avslöjar hur läkemedelsmolekyler interagerar med sitt målprotein och ändrar proteinets konformation, eller form, som bestämmer dess funktion.
Dock, dessa förutsägelsemöjligheter fungerar ännu inte i tillräckligt stor skala eller tillräckligt hög hastighet för att läkemedelsföretag ska kunna använda dem i sin utvecklingsprocess.
"Att översätta dessa framsteg i prediktiv noggrannhet till att påverka industriellt beslutsfattande kräver att i storleksordningen 10, 000 bindande affiniteter beräknas så snabbt som möjligt, utan förlust av noggrannhet, ", sa Jha. "Att producera snabba insikter kräver en beräkningseffektivitet som bygger på utvecklingen av nya algoritmer och skalbara mjukvarusystem, och smart allokering av superdatorresurser."
Jha och hans medarbetare vid Rutgers University, där han också är professor vid avdelningen för elektro- och datateknik, och University College London designade ett mjukvaruramverk för att stödja korrekt och snabb beräkning av bindningsaffiniteter samtidigt som användningen av beräkningsresurser optimerades. Detta ramverk, kallas High-Throughput Binding Affinity Calculator (HTBAC), bygger på RADICAL-Cybertools-projektet som Jha leder som huvudutredare för Rutgers Research in Advanced Distributed Cyberinfrastructure and Applications Laboratory (RADICAL). Målet med RADICAL-Cybertools är att tillhandahålla en uppsättning mjukvarubyggstenar för att stödja arbetsflödena i storskaliga vetenskapliga applikationer på högpresterande datorplattformar, som samlar beräkningskraft för att lösa stora beräkningsproblem som annars skulle vara olösliga på grund av den tid som krävs.
Inom datavetenskap, arbetsflöden hänvisar till en serie bearbetningssteg som krävs för att slutföra en uppgift eller lösa ett problem. Speciellt för vetenskapliga arbetsflöden, det är viktigt att arbetsflödena är flexibla så att de dynamiskt kan anpassas under körning för att ge de mest exakta resultaten samtidigt som den tillgängliga beräkningstiden utnyttjas effektivt. Sådana adaptiva arbetsflöden är idealiska för läkemedelsupptäckt eftersom endast läkemedel med hög bindningsaffinitet bör utvärderas ytterligare.
"Den avvägning som önskas mellan den erforderliga noggrannheten och beräkningskostnaden (tid) förändras under hela läkemedlets upptäckt när processen går från screening till val av leads och sedan leadoptimering, ", sade Jha. "Ett betydande antal föreningar måste screenas billigt för att eliminera dåliga bindemedel innan mer exakta metoder behövs för att urskilja de bästa bindemedlen. För att tillhandahålla den snabbaste tiden till lösning krävs övervakning av simuleringarnas framsteg och baserat beslut om fortsatt utförande på vetenskaplig betydelse."
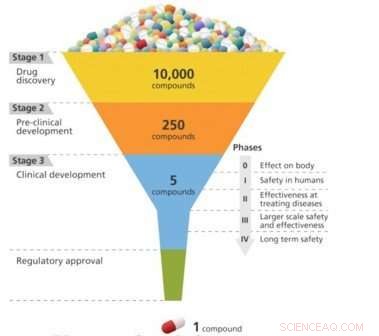
Ett schema över läkemedelsutvecklingsprocessen, som successivt finslipar de mest effektiva kandidaterna från en stor initial pool. Kredit:Brookhaven National Laboratory
Med andra ord, det skulle inte vara meningsfullt att fortsätta simuleringar av en viss läkemedel-proteininteraktion om läkemedlet binder proteinet svagt jämfört med de andra kandidaterna. Men det skulle vara vettigt att allokera ytterligare beräkningsresurser om ett läkemedel visar en hög bindningsaffinitet.
Att stödja adaptiva arbetsflöden i stor skala som är typiska för läkemedelsupptäcktsprogram kräver avancerade beräkningsmöjligheter. HTBAC tillhandahåller sådant stöd genom ett flexibelt mellanprogramvarulager som möjliggör adaptiv exekvering av algoritmer. För närvarande, HTBAC stöder två algoritmer:förbättrad sampling av molekylär dynamik med approximation av kontinuumlösningsmedel (ESMACS) och termodynamisk integration med förbättrad sampling (TIES). ESMACS, en beräkningsmässigt billigare men mindre rigorös metod än TIES, beräknar bindningsstyrkan för ett läkemedel till dess målprotein på basis av simuleringar av molekylär dynamik. Däremot TIES jämför de relativa bindningsaffiniteterna för två olika läkemedel till samma protein.
"ESMACS tillhandahåller en snabb kvantitativ metod som är tillräckligt känslig för att bestämma bindningsaffiniteter så att vi kan eliminera dåliga bindemedel, medan TIES ger en mer exakt metod för att undersöka bra bindemedel eftersom de förfinas och förbättras, " sa Jumana Dakka, en andraårs Ph.D. student vid Rutgers och medlem i RADICAL-gruppen.
För att bestämma vilken algoritm som ska köras, HTBAC analyserar bindningsaffinitetsberäkningarna vid körning. Denna analys informerar beslut om antalet samtidiga simuleringar som ska utföras och om stimuleringssteg ska läggas till eller tas bort för varje undersökt läkemedelskandidat.
Att sätta ramarna på prov
Jha's team demonstrated how HTBAC could provide insight from drug candidate data on a short timescale by reproducing results from a collaborative study between University College London and the London-based pharmaceutical company GlaxoSmithKline to discover drug compounds that bind to the BRD4 protein. Known to play a key role in driving cancer and inflammatory diseases, the BRD4 protein is a major target of bromodomain-containing (BRD) inhibitors, a class of pharmaceutical drugs currently being evaluated in clinical trials. The researchers involved in this collaborative study are focusing on identifying promising new drugs to treat breast cancer while developing an understanding of why certain drugs fail in the presence of breast cancer gene mutations.
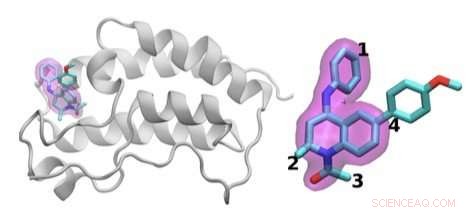
The scientists investigated the chemical structures of 16 drugs based on the same tetrahydroquinoline (THQ) scaffold. On the left is a cartoon of the BRD4 protein bound to one of these drugs; on the right is a molecular representation of a drug with the THQ scaffold highlighted in magenta. Regions that are chemically modified between the drugs investigated in this study are labeled 1 to 4. Typically, only a small change is made to the chemical structure of one drug to the next. This conservative approach makes it easier for researchers to understand why one drug is effective, whereas another is not. Credit:Brookhaven National Laboratory
Jha and his team concurrently screened a group of 16 closely related drug candidates from the study by running thousands of computational sequences on more than 32, 000 computing cores. They ran the computations on the Blue Waters supercomputer at the National Center for Computing Applications, University of Illinois at Urbana-Champaign.
In a real drug design scenario, many more compounds with a wider range of chemical properties would need to be investigated. The team members previously demonstrated that the workload management layer and runtime system underlying HTBAC could scale to handle 10, 000 concurrent tasks.
"HTBAC could support the concurrent screening of different compounds at unprecedented scales—both in the number of compounds and computational resources used, " said Jha. "We showed that HTBAC has the ability to solve a large number of drug candidates in essentially the same amount of time it would take to solve a smaller set, assuming the number of processors increases proportionally with the number of candidates."
This ability is made possible through HTBAC's adaptive functionality, which allows it to execute the optimal algorithm depending on the properties of the drugs being investigated, improving the accuracy of the results and minimizing compute time.
"The lead optimization stage usually considers on the order of 10, 000 small molecules, " said Jha. "While experiment automation reduces the amount of time needed to calculate the binding affinities, HTBAC has the potential to cut this time (and cost) by an order of magnitude or more."
With HTBAC, TIES requires approximately 25, 000 central processing unit (CPU) core hours for a single prediction. At least a 250 million core hours would be needed for a large-scale study to support a pharmaceutical drug screening campaign, with a typical turnaround time of about two weeks. HTBAC could facilitate running studies requiring sustained usage of millions of core hours per day.
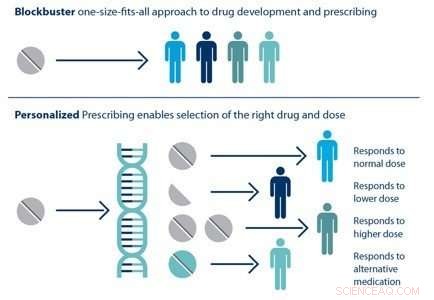
Individual patients respond differently to drugs. The ability to predict which treatment is best for a particular patient based on his or her genetic sequence is the goal of personalized medicine. Credit:Brookhaven National Laboratory
When the University of College London–GlaxoSmithKline study concludes, Jha and his team hope to be given the experimental data on the tens of thousands of drug candidates, without knowing which candidate ended up being the best one. Med denna information, they could perform a blind test to determine whether HTBAC provides an improvement in compute time (for a given accuracy) over the existing automated methods for drug discovery. Om nödvändigt, they could then refine their methodology.
Applying scalable computing to precision medicine
HTBAC not only has the potential to improve the speed and accuracy of drug discovery in the pharmaceutical industry but also to improve individual patient outcomes in clinical settings. Using target proteins based on a patient's genetic sequence, HTBAC could predict a patient's response to different drug treatments. This personalized assessment could replace the traditional one-size-fits-all approach to medicine. Till exempel, such predictions could help determine which cancer patients would actually benefit from chemotherapy, avoiding unnecessary toxicity.
According to Jha, the computation time would have to be significantly reduced in order for physicians to clinically use HTBAC to treat their patients:"Our grand vision is to apply scalable computing techniques to personalized medicine. If we can use these techniques to optimize drugs and drug cocktails for each individual's unique genetic makeup on the order of a few days, we will be empowered to treat diseases much more effectively."
"Extreme-scale computing for precision medicine is an emerging area that CSI and Brookhaven at large have begun to tackle, " said CSI Director Kerstin Kleese van Dam. "This work is a great example of how technologies we originally developed to tackle DOE challenges can be applied to other domains of high national impact. We look forward to forming more strategic partnerships with other universities, pharmaceutical companies, and medical institutions in this important area that will transform the future of health care."














